











成果简介
金属-空气电池也因其理论能量密度高、电池性能好、放电电压稳定等优点而备受关注。燃料电池和金属-空气电池都可以等温将储存在燃料或氧化剂中的化学能转化为电能,本质上是一种氧化还原反应。氧电极反应包括充电过程中的OER和放电过程中的ORR两种反应模式,是这些能量装置的关键步骤。VC具有良好的金属性能,在催化析氧反应过程中,VC可以在酸性和碱性化学环境中稳定存在。在实际应用中具有超高的稳定性和耐腐蚀性,可以抵抗反应中的“催化剂中毒”。VC虽然具有许多优异的性能,但对其电催化性能的系统理论研究还有待完善。
宜宾职业技术学院陈泽华和河南理工大学邓超等人通过第一性原理计算研究了VC对ORR的催化性能。首先,本文考虑了ORR的电子性质、吸附能和可能的反应途径。通过系统阐述VC表面不同吸附方式对O2在不同吸附位点的吸附强度,本文选择了两种具有优异吸附稳定性和电荷转移的结构继续进行ORR路径研究。本文研究结果为改进燃料电池ORR动力学的新型催化剂的设计提供有益的指导。
计算方法
本文基于自旋极化密度泛函理论(DFT),使用Vienna ab initio simulation package (VASP)进行第一性原理计算。广义梯度近似(GGA)中的Perdew-Burke-Ernzerhof (PBE)泛函用于DFT计算中的描述交换函数和相关函数,电子-离子相互作用用投影增强波(PAW)伪势来描述。所有的计算都基于450 eV的平面波截止和10−5以内的能量收敛准则,每个原子上的力为0.01 eV/Å,并在所有VC表面构建了一个3 × 3 × 1的超级单体(含16个钒原子和16个碳原子),沿z方向增加真空厚度15 Å,该真空厚度足够大,可以忽略簇与周期图像之间的相互作用。本文利用8 × 8 × 8 Å超级单体模拟了反应过程中吸附的分子(O2),K点采样采用蒙霍斯特-包(Monkhorst-Pack,MP)方案,采用布里渊区积分,其中采用5 × 5 × 1的MP网格进行几何优化和电子分析,采用Grimme半经验色散校正方法(DFT-D3)来说明范德华相互作用,整个计算基于自旋极化。
结果与讨论
图1(a)和(b)为VC结构的俯视图和侧视图,VC的晶格常数为a = 10.09 Å,b = 11.47 Å,V-C键长为2.0 Å,本文的结果与前人的结果吻合较好。本文在VC晶体结构基面上选择了四个可能的ORR位点,分别是外V原子的顶部位置(位点1)、中间C原子的顶部位置(位点2)、C原子与V原子之间的桥接位置(位点3)和C原子与V原子之间的空心位置(位点4)[见图1a]。原始VC的电子能带结构和总态密度(DOS)如图1b所示。计算的原始VC的DOS继续接近费米能级,价带的顶部和导带的底部分别来自V的d轨道和C的p轨道。另外,从0 eV到3 eV的DOS主要由金属原子控制。由于与碳轨道的杂化,电子将从低能级跃迁到高能级,导致价带与部分填充的导带重叠。综上所述,上述理论分析结果表明,原始VC具有良好的金属性能,有利于ORR催化过程中基底材料与吸附质之间的电荷转移。
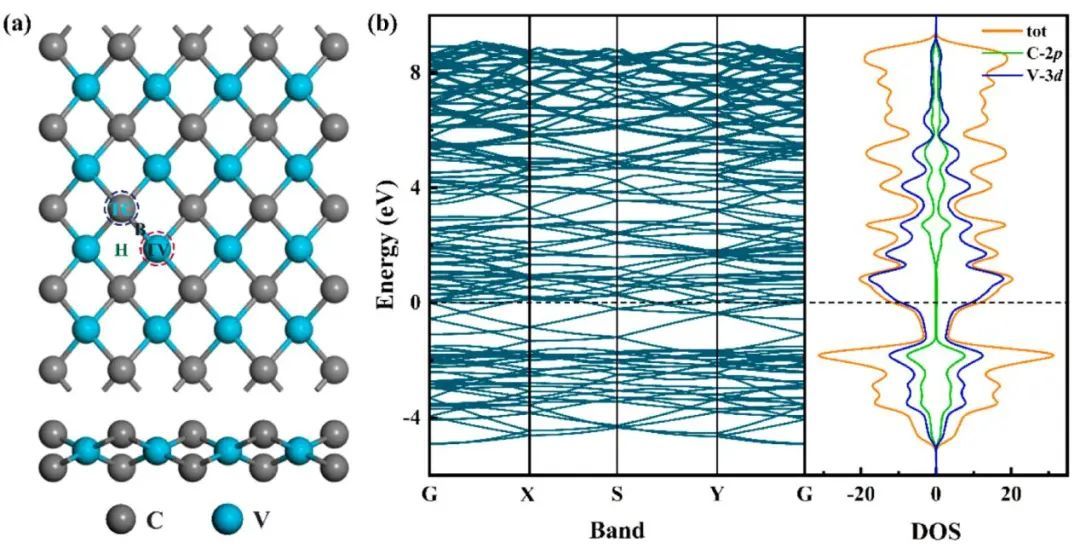
图1 (a)原始VC结构示意图的俯视图和侧视图,以及原始VC表面的四个ORR反应位点;(b)原始VC各元素的能带结构和态密度(DOS),其中费米能级为零(虚线)。
ORR的前提是对O2的吸附,而吸附结构和吸附方法有不同的后续反应路径,因此对应不同的反应。同时,O2在底物上的稳定吸附是必不可少的,因为它可以直接影响后续的碱性反应步骤。为了研究VC催化剂的ORR催化活性,本文首先研究了VC表面不同吸附位点端对和侧对吸附O2分子的吸附强度。由于VC结构表面相当复杂,不同吸附位点的吸附强度可由吸附位点的电子构型、原子半径、电子亲和、配位场和配位环境等多种因素决定。较小的吸附能表明O2分子能在VC表面积极吸附,具有较高的稳定性。负Eads表明O2分子的吸附是一个放热过程。侧面吸附在空心位点上的O2分子的Eads为-4.63 eV(如表1所示),并且在4个选择的位点中吸附能为负,因此侧面吸附的O2分子更倾向于吸附在空心位点上。此外,端对端吸附在VC表面的四种构型中,H位点的吸附能负得最大,为- 3.35 eV(见表1),吸附效果最好且更稳定。然而,这种构型的优化结构与侧对吸附选择的最优构型非常相似。为了避免重复研究,本文选择了另外两种具有同样优异吸附能的构型,即O2分子分别吸附在桥位和V原子顶部。虽然这两种构型的吸附能相似,但在V原子顶部的吸附效果更好。因此,本文选择该构型来研究其ORR催化活性。
表1 O2分子在纯VC上的吸附能(Eads)、催化剂上O-O键的距离范围(ΔdO-O)、底物到活化O2分子的电荷转移数(ΔQ)。
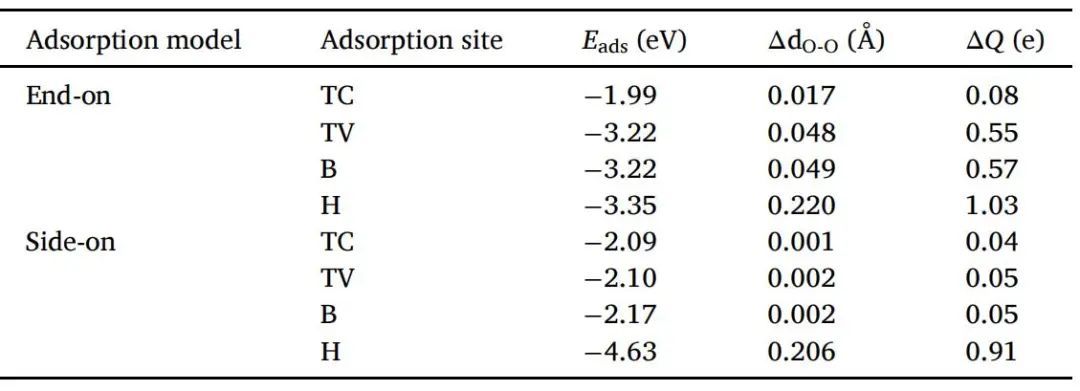
为了进一步了解Ead-TV和Sad-H这两种构型的稳定性,本文还进行了电荷密度差和bader电荷分析,研究O2分子与VC表面之间的电荷转移。电荷密度差分析(如图2所示)表明,VC表面向O2分子有明显的电荷转移,VC表面失去电子,成为电子供体。相反,O2分子得到了电子,这与之前的研究结果一致。根据Bader电荷分析(如表1所示),正的ΔQ表示电荷从子层转移到O层,其中V原子贡献最大。对于Sad-H,从VC表面到O2分子的电子转移量约为0.91 e,这是很大的O2分子侧对VC表面的吸附比其他三种构型更大。对于Ead-TV,电荷转移量约为0.55 e。
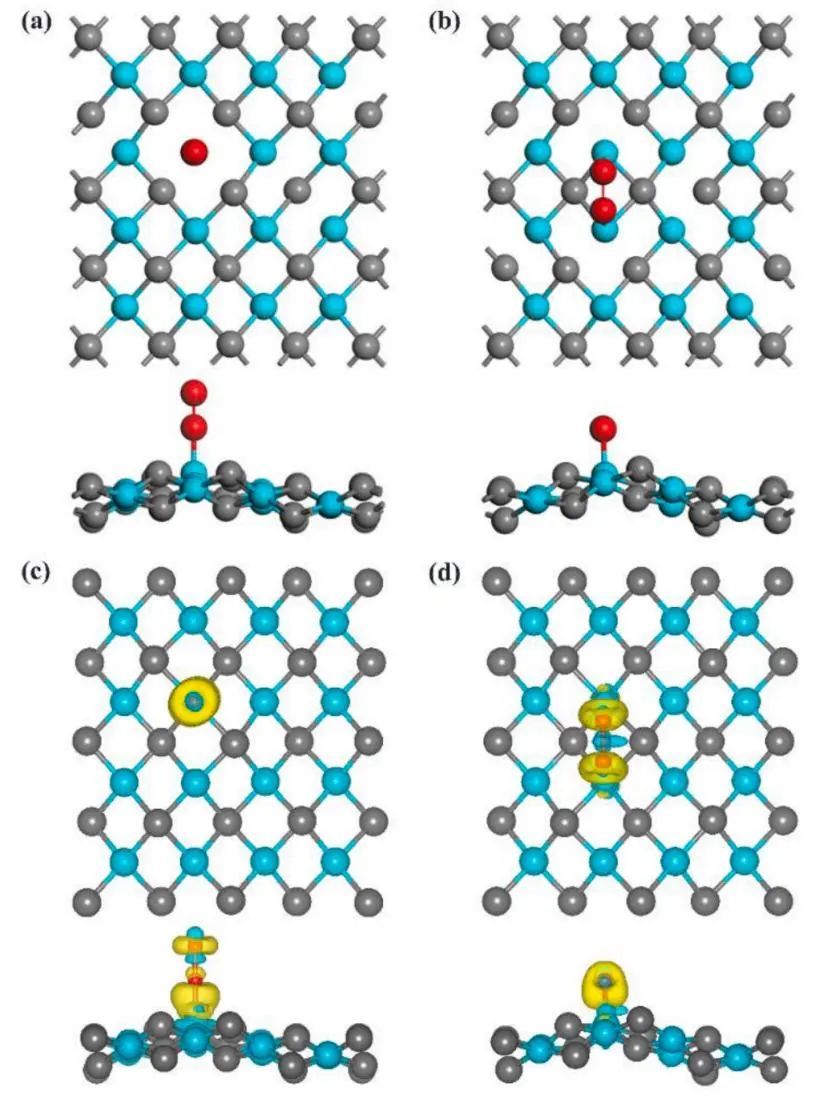
图2 (a-b) VC表面氧端对和侧对最优吸附位点的几何构型;(c-d) Ead-TV与Sad-H的电荷密度差。
此外,为了进一步了解O2分子在VC表面稳定吸附的内部机理,本文计算了O原子上的p轨道投影、V原子的d轨道和C原子p轨道上自旋极化的DOS(如图3所示),以及O2分子与VC底物的相互作用。发现O-2p轨道与V-3d轨道和C-2p轨道之间存在明显的轨道杂化,这在O2分子与VC底物的相互作用中起着至关重要的作用。而且,这两种构型具有良好的磁性,主要来自于V的三维态,这是由于不对称自旋向上和向下的通道,杂质水平也有利于电子转移带隙的减小。同时,从PDOS也可以看出,VC未填充的d轨道接受电子形成成键状态,增强了对O2的吸附。另外,VC的d轨道为O2的2π*和3σ轨道提供电子。反键电子的增强导致O-O键的延伸和活化,键长增加幅度为0.001⁓0.220 Å。因此,可以提高这些催化剂的导电性,从而进一步提高电催化效率。
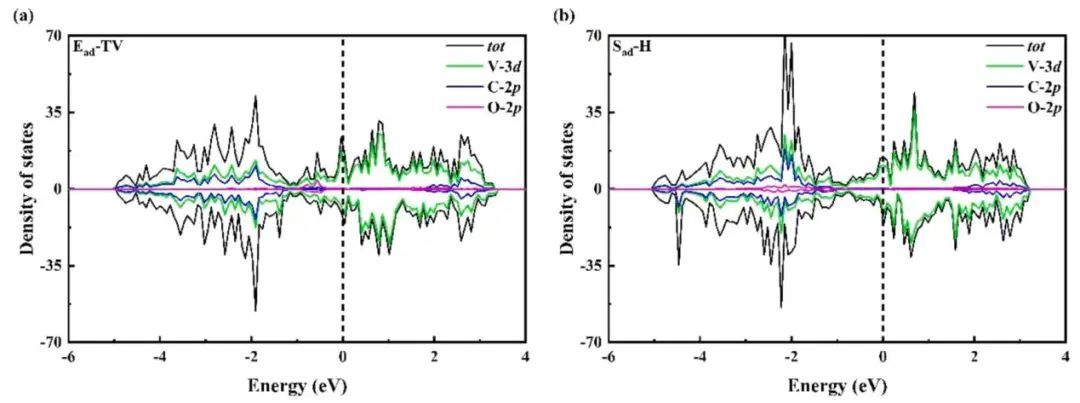
图3 (a)Ead-TV的DOS;(b)Sadh的DOS。费米能量设置为零(虚线)。
一般认为,吸附质的化学吸附能和催化剂的表面能与两个因素有关:活性中心与吸附质的轨道重叠程度和表面-吸附剂相互作用的反键态填充程度。前者可以在一定程度上用pz中心来表示,而后者可以用投影晶体轨道汉密尔顿居群(pCOHP)来量化。图4为分离态成键态与反键态的相互作用,端对端吸附在V的顶部,侧对端吸附在O2的中空位点。从COHP图可以看出,对于Ead-TV构型,O2中的1π轨道提供了大量的电子,是O-O键的主要供体。
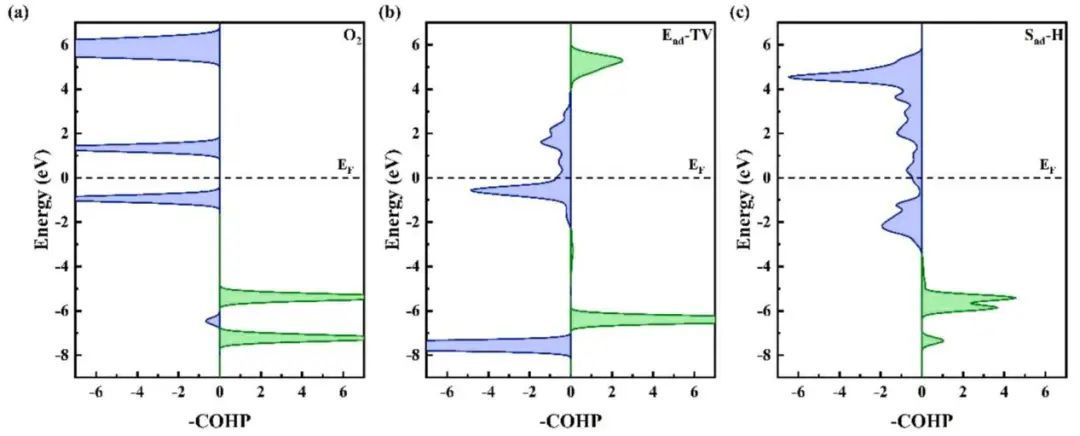
图4 O2 (a)分离态的COHP;(b)端对吸附在V顶部,(c)侧对吸附在空心处。COHP中的成键态和反键态分别用绿色和蓝色表示。
要了解ORR的反应机理,第一步也是最重要的一步是确定两电子和四电子途径中哪一种在能量上更有利。这两种途径的主要区别在于最终产物是过氧化氢(H2O2)还是水(H2O)。在燃料电池方面,更理想的方式是通过四电子路径产生H2O,这种方式可以充分利用O2,并且必然会输出更高的电压,从而获得更大的应用潜力。通过计算过氧化氢在VC底物上的吸附性能,确定了ORR的反应路径。随着O-O键的断裂,H2O2在Ead-TV上倾向于自发分裂成O* + H2O,在Sad-H上倾向于自发分裂成OH* + OH*,而不是作为稳定分子吸附在底物上。因此,本文主要研究ORR在VC底物上的四电子反应路径。如图5所示,本文总结了VC底物上可能的ORR反应途径,包括两种反应机制:解离(O2直接解离成两个O*种或OOH*直接解离成O* + H2O种)和结合(O2被氢化成OOH*种或OOH*被氢化成HOOH*种)。与O2解离反应相比,O2加氢反应的能垒相对较低。
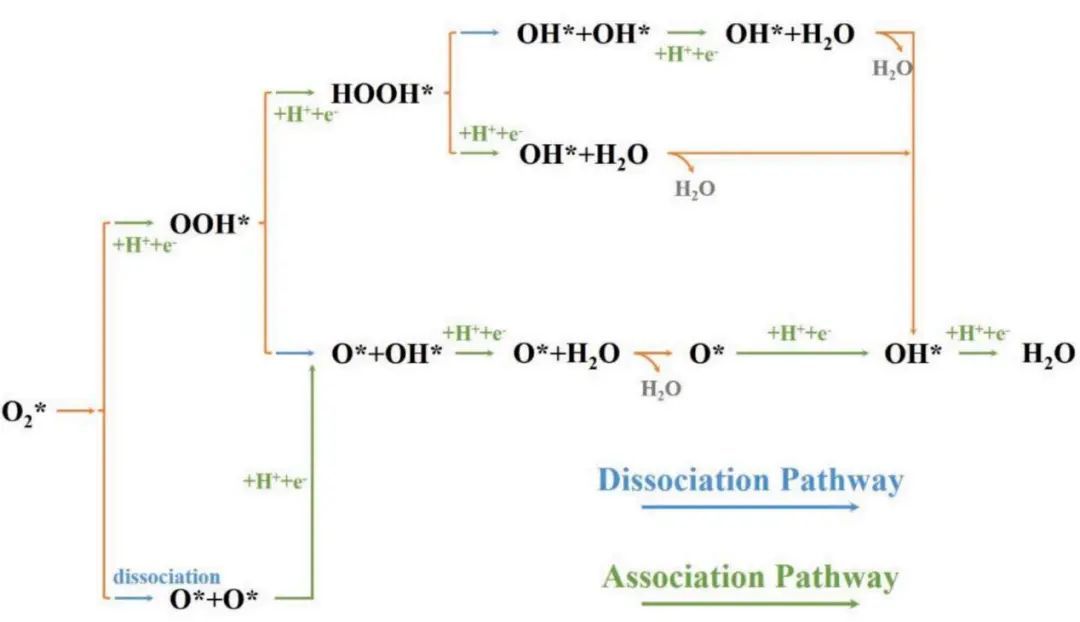
图5 O2还原为H2O的可能反应路径图。
图6(a)和(c)分别为整个ORR反应在Ead-TV和Sad-H上的四电子路径和中间构型示意图。在O2在活性中心吸附的基础上,O2首先被H+/e−氢化成OOH*(*表示吸附在催化剂表面的中间体)。OOH*得到H+/e−对,生成第一个H2O分子和O*,然后第一个H2O分子从表面释放出来。O*与H+/e−对反应生成OH*,最后OH*与H+/e-对结合生成第二个H2O分子。详细的ORR四电子路径如图(3a)-3d所示。图6(c)和(d)分别显示了在Ead-TV和Sad-H上发生的ORR的自由能谱。结果表明:对于Ead-TV和Sad-H,在U = 0 V时,4e机制中所有中间体形成的反应步骤都是下坡的,是放热的,可以自发进行;为了更好地确定ORR反应过程中各步骤的难度,本文采用电位测定步骤(PDS)来评价反应势垒,即两个吸附反应中间体在最大热力学势(U = 1.23 V)下的最大吉布斯自由能。

图6 (a,b) O2在VC底物上端对端吸附的四电子路径各步的路径图和相应的自由能谱;(c,d) O2侧边吸附在VC底物上的四电子路径的每一步的路径图和相应的自由能谱。
为了评价电催化反应的性能,本文计算了0 V电极电位下Ead-TV和Sad-H的ORR的ΔG值和理论过电位(ηORR)。ORR引起的总ΔG为−4.92 eV,由ORR热力学决定,即2G(H2) + G(O2) - 2G(H2O) = 4.92 eV。理论过电位由四个电化学步骤的最大自由能差决定(ΔG)。计算结果表明,Ead-TV和Sad-H的过电位分别为0.93 V和0.81 V。
通过比较,本文发现,与Ead-TV相比,本文选择的两种结构中Sad-H的过电位相对较小,因此其催化活性也很出色。研究发现,虽然这两种构型在纯Pt(111)表面的过电位略高于0.45 V,在PtHg4(1 1 0)表面的过电位略高于0.46 V,但与含贵金属的Pt基催化剂相比,本文选择的构型应用成本更低,结皮中含量更丰富,因此商业价值更高。同时,这两种构型的过电位均低于FeN3-Gra的1.01 V,甚至Sad-H构型也低于或相当于RhN3-Gra的0.90 V和CoN3-Gra的0.83 V。这表明,从热力学的角度来看,Ead-TV和Sad-H的ORR催化活性与这些电催化剂相当甚至更好。
结论与展望
在这项工作中,本文首先通过第一性原理计算研究了本征VC的结构、电子和磁性能,发现本征VC具有金属性质,因此具有良好的导电性,可以作为电子给体。结果表明,VC表面能稳定吸附O2,且Ead-TV和Sad-H两种构型吸附效果较好。最后,本文研究了ORR在Ead-TV和Sad-H上的催化性能和反应机理。四电子反应机制比二电子反应机制更有利,因为HOOH*中间体不能稳定吸附在这两种催化剂上。虽然ORR反应过程中的过电位比较接近,但Sad-H构型的值略低,其催化活性与传统的工业催化剂相当甚至更好。综上所述,本文的研究结果不仅提供了一种新的可能的ORR电催化剂,而且为其他ORR催化剂的最佳活性中心的选择提供了一种新的策略。
文献信息
Lin, L., Yang, X., Shi, P., Yan, L., Xie, K., Deng, C., & Chen, Z. (2023). Probing the Origin of Transition Metal Carbide VC for Oxygen Reduction Reaction: A DFT Study. Surfaces and Interfaces, 103100.
ht+tps://doi.org/10.1016/j.surfin.2023.103100


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









