










 文章介绍了新疆大学研究人员通过掺杂IIIA-VIA族元素(如Sb和Ga)到SnS2单层的S缺陷位置,利用DFT计算筛选出的高效HER催化剂。Sb/Ga掺杂提高了SnS2的导电性和催化活性,通过分析能带结构和氢吸附行为,揭示了其在氢进化反应中的优势。
文章介绍了新疆大学研究人员通过掺杂IIIA-VIA族元素(如Sb和Ga)到SnS2单层的S缺陷位置,利用DFT计算筛选出的高效HER催化剂。Sb/Ga掺杂提高了SnS2的导电性和催化活性,通过分析能带结构和氢吸附行为,揭示了其在氢进化反应中的优势。

二硫化锡(SnS2)是用于析氢反应(HER)的有效非贵金属催化剂。然而,SnS2在电催化HER中的实际应用受到较差导电性和催化性能的阻碍。在此,新疆大学崔秀花、吴荣等人通过在S缺陷SnS2单层上掺杂14种主族元素,并利用密度泛函理论(DFT)计算筛选出最具潜力的HER催化剂。
计算方法
作者使用维也纳从头算模拟包(VASP)进行第一性原理计算,并采用了投影增强波(PAW)方法和广义梯度近似中的Perdew-Burke-Ernzerhof(PBE)泛函,以及使用Grimme的DFT-D2方法来描述范德华(vdw)相互作用。作者建立了一个在z方向上具有15Å真空层的4×4×1 2H–SnS2超胞平板模型,其中含有32个S原子和16个Sn原子。作者采用以Γ为中心的2×2×1(用于结构优化)和5×5×1(用于电子结构计算)K点网格对布里渊区进行采样。作者将平面波的截止能量设置为500eV,并且采用0.05 eV的高斯弥散来加速收敛。在结构优化过程中,作者将所有原子保持驰豫,直到总能量和赫尔曼-费曼力的收敛标准分别达到1.0×10−5 eV/atom和0.01 eV/Å。此外,作者在整个计算过程中还考虑了自旋极化,以及利用正则(NVT)系综进行从头算分子动力学(AIMD)模拟。
结果与讨论
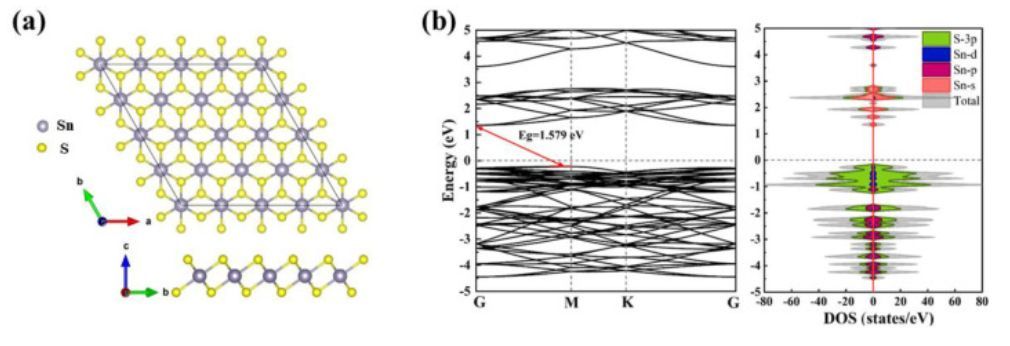
图1 SnS2模型结构、能带结构和态密度
如图1所示,SnS2的晶格参数为a=b=3.688Å,c=5.906Å,Sn–S键长为2.590Å。此外,作者计算了SnS2单层的能带结构、总态密度(TDOS)和投影态密度(PDOS),具体如图1b所示。结果表明,SnS2单层具有1.579eV的间接带隙,其中价带最大值(VBM)位于M点附近,而导带最小值位于Γ点。而SnS2单层的VBM主要由S-3p轨道贡献,而CBM主要由杂化的S-3p和Sn-5s态组成。
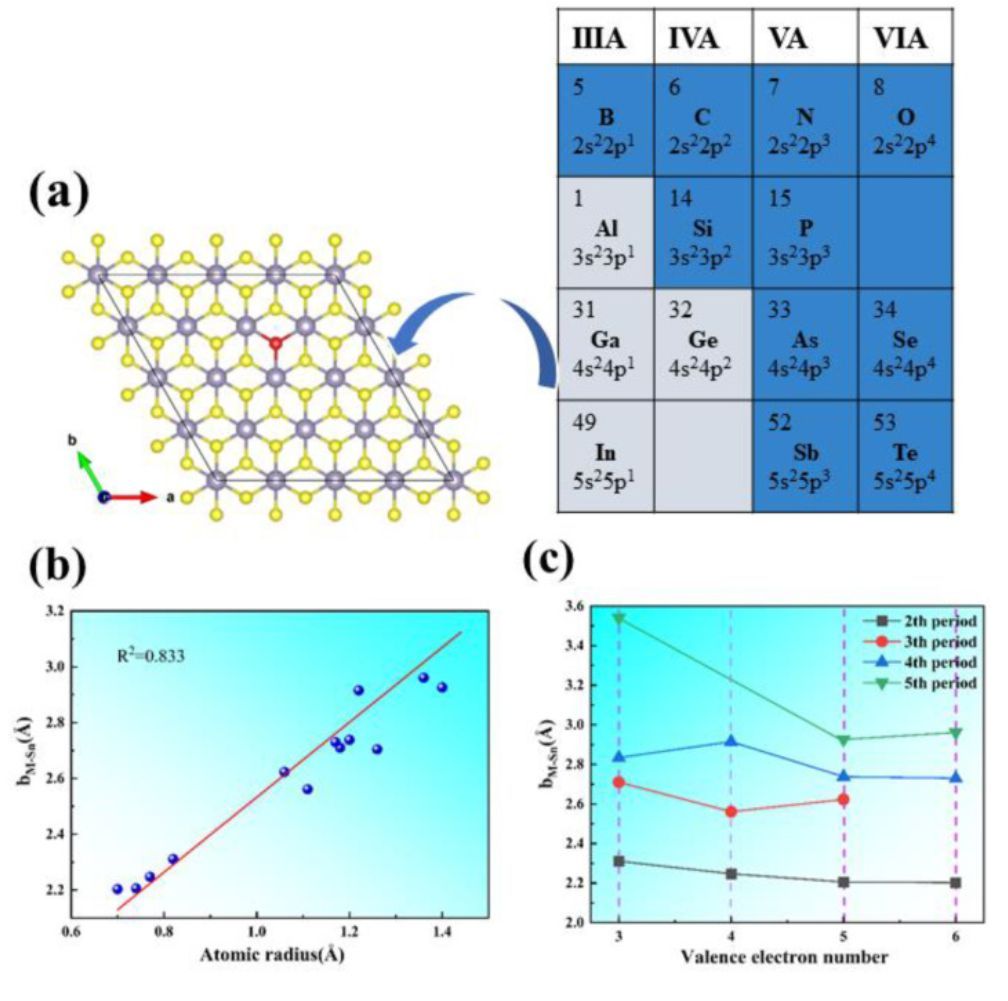
图2 IIA-VIA族原子掺杂Vs-SnS2示意图、bM-Sn与掺杂原子半径以及价电子数目的关系
由于纯SnS2的HER催化性能较差,作者从14种掺杂结构中依次选择了几个不同的IIIA-VIA族原子掺杂到纯SnS2基面上的S缺陷位置(红球的位置如图2a所示)(M@SnS2,M为掺杂原子),掺杂浓度为3.13%。如图2b和2c所示,M原子的原子半径与M−Sn的键长呈线性相关,即M原子半径越大,M-Sn的键长就越长。因此,在类似的主族中,bM-Sn的值随着M原子序数的增加而增加。
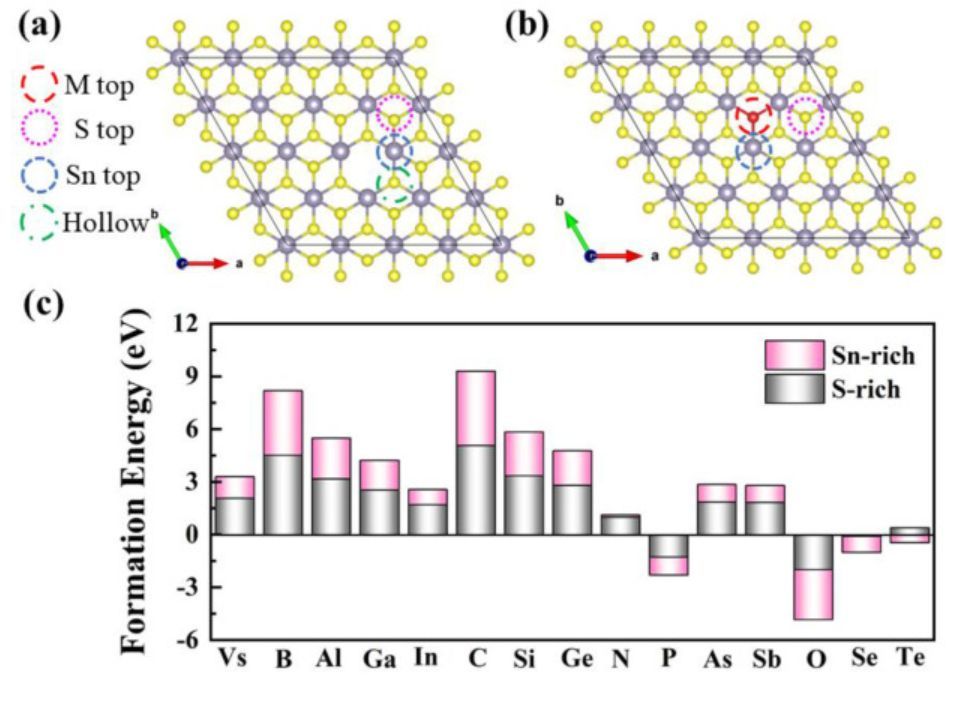
图3 氢吸附位点和形成能
氢吸附过程是HER的第一步,可以用吸附能来描述,并且H倾向于吸附在具有最低吸附能的位点上。对于Vs-SnS2,作者比较了三个可能的吸附位点,即暴露Sn原子的顶部位点、S缺陷周围的S原子的顶部位置和中空位点(见图3a)。对于M@SnS2,作者研究了三个可能的吸附位点,包括掺杂原子(M)的顶部位点、Sn原子的顶部位点和相邻S原子的顶部位置(见图3b)。对于Vs-SnS2,富S和富Sn的形成能分别为2.076eV和1.225eV,表明在富Sn条件下更容易获得S空位。如图3c所示,形成能与掺杂元素的电负性负相关,Ef(O) <Ef(Se)<Ef(Te),Ef(In)<Ef(Ga)<Ef(Al)。因此,具有更大电负性的分子更容易吸附在基底上。
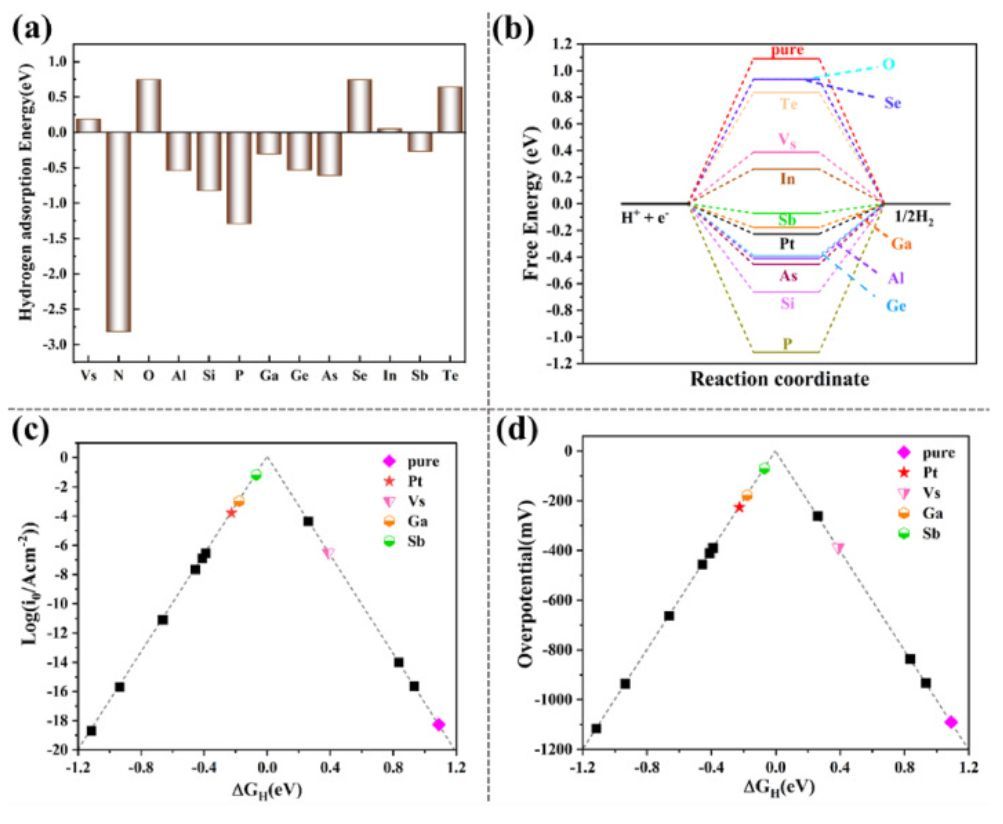
图4 吸附能、势能面和火山曲线
在Vs-SnS2中,H原子优先吸附在S顶位上,而吸附在Sn顶位和中空位点上的H原子倾向于向相邻的S位点移动。至于M@SnS2,H原子倾向于吸附在O/Se/Te/Sb/In@SnS2相邻S原子的顶位,而对于N/Al/Si/P/Ga/Ge/As@SnS2,掺杂剂的顶位是最稳定的吸附位。此外,所有Sn顶位的H原子倾向于移动到掺杂剂顶位或相邻S原子的顶位。图4(a)显示了每种构型中最稳定的氢吸附位点对应的吸附能,其中除O/Se/Te硫族元素外,主族元素掺杂原子可以提高Vs-SnS2的氢吸附能。如图4(b)所示,与Vs-SnS2相比(ΔGH=0.388eV),Sb@SnS2和Ga@SnS2具有最佳的HER催化活性,相应的ΔGH分别为-0.070 eV和-0.178 eV。此外,在图4(c)和(d)中,Sb@SnS2和Ga@SnS2位于Vs-SnS2上方,更靠近火山曲线的峰值。
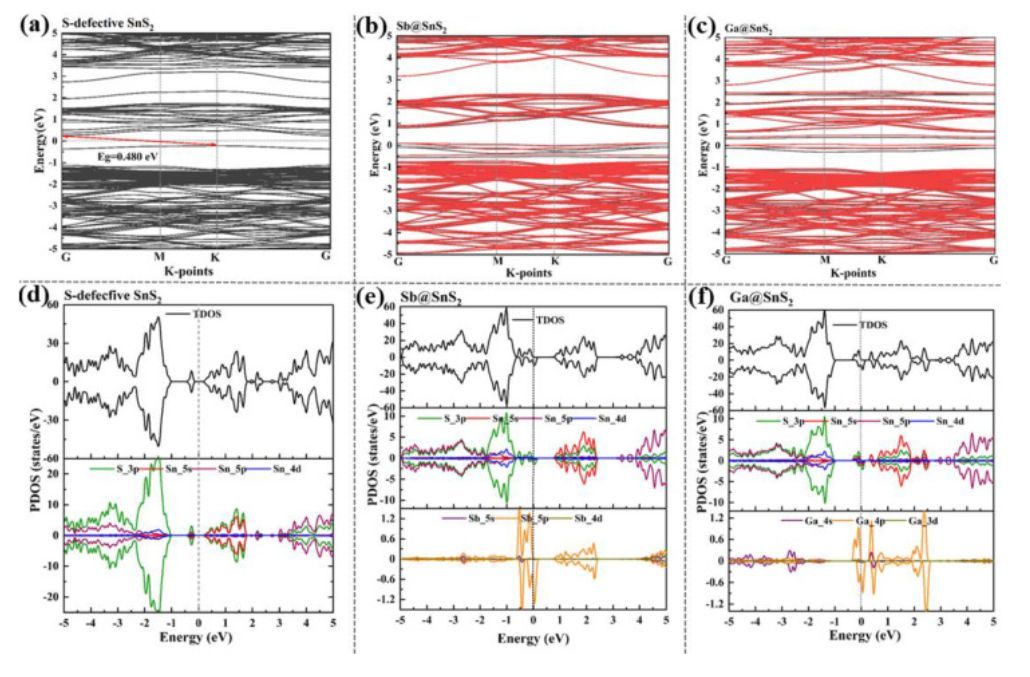
图5 能带结构和PDOS
为了深入研究M@SnS2(M=Sb,Ga),作者分析了M@SnS2的能带结构和投影态密度(PDOS)(图5(b)(c)(e)(f)),并与纯SnS2单层(图1b)和S缺陷SnS2(图5(a)和(d))进行比较。从图中可以看出,与纯SnS2(Eg=1.579eV)相比,Vs-SnS2(Eg=0.48eV)的带隙显著减小,杂质能级更接近CBM,表明S空位是SnS2的n型掺杂。此外,作者发现Vs-SnS2系统是非自旋极化的。S空位的引入并没有改变SnS2中CBM和VBM的组成,即VBM仍然主要来源于S-3p态,而CBM主要由S-3p和Sn-5s态杂化。而新形成的杂质态是由S-3p贡献的。尽管Vs-SnS2的带隙比纯SnS2低得多,但在费米能级附近没有合适的未占据态,这导致其与H自由基不可实现键合。然而,在Vs-SnS2中引入Sb和Ga可以进一步将本征SnS2的带隙降低到0eV(图5(b)和(c)),从而显著改善了SnS2的导电性质。图5(e)显示了Sb@SnS2的能级下移,通过Sb取代引入接近VBM的杂质态,表明其是p型掺杂。对于纯SnS2表面,H电子填充的第一个可用态是CBM,其比价带最大值(VBM)高约1.579eV(图1(b)),导致H-s轨道和S-p轨道之间的杂化较弱。相反,Sb掺杂在费米能级处产生部分填充的S-3p态,并且很容易地容纳H电子以产生H–S结合。从图5(f)中可以看出,由于Ga原子的引入,在费米能级周围出现了几个新的态,这进一步缩短了CBM和费米能级之间的间隙。
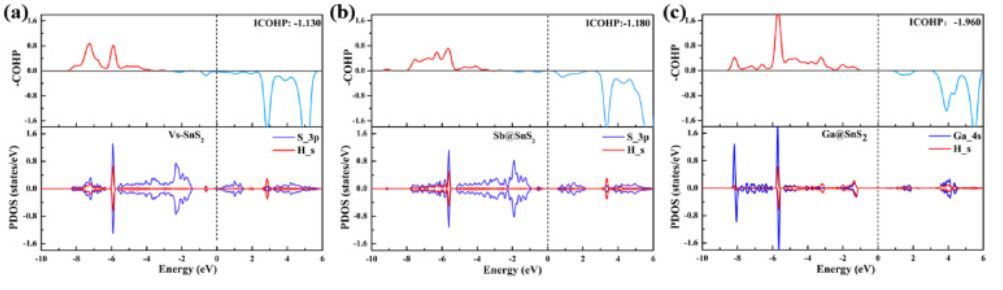
图6 COHP
如图6(a)~(c)所示,成键态可以基于正重叠布居(-pOHP>0)来表征,而负重叠布居则表征反键态。与Vs-SnS2系统的COHP相比,Sb@SnS2系统中的反键态填充减少,并且Ga@SnS2系统的反键态几乎为空。因此,Sb/Ga@SnS2系统中M(M=S,Ga)和H原子之间的相互作用更强。ICOHP结果如下:Vs-SnS2(−1.130)>Sb@SnS2(−1.180)>Ga@SnS2(-1.960),这表明Ga/Sb@SnS2比Vs-SnS2具有更强的H自由基键合能力。
结论与展望
研究发现,锚定在S缺陷SnS2基面上的Sb/Ga原子具有优异的HER性能,相应的ΔGH分别为−0.07和−0.178 eV,这比铂(Pt)催化剂的性能更好。作者通过态密度(DOS)分析发现,催化性能的提高归因于Sb/Ga原子的引入,其增强了原始SnS2表面的导电性,并提供了费米能级附近的未占据态,从而有效降低了H自由基和M@SnS2表面之间电荷转移的能垒。该工作揭示了一种提高SnS2基面HER活性的有效策略。
文献信息
Meiling Pan et.al Enhancing the hydrogen evolution reaction by group IIIA-VIA elements doping in SnS2 basal plane International Journal of Hydrogen Energy 2023
ht+tps://doi.org/10.1016/j.ijhydene.2023.07.220


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









