










 南开大学的研究者探讨了电化学电势如何影响氮掺杂石墨烯衬底上的单原子催化剂(SACs)中“前沿轨道”,发现电势变化导致吸附能反转,这对于理解金属单原子催化剂在电化学条件下的催化机制具有重要意义。
南开大学的研究者探讨了电化学电势如何影响氮掺杂石墨烯衬底上的单原子催化剂(SACs)中“前沿轨道”,发现电势变化导致吸附能反转,这对于理解金属单原子催化剂在电化学条件下的催化机制具有重要意义。


电子结构对于理解金属单原子催化剂(SACs)的催化机理至关重要,尤其是在电化学条件下。南开大学刘锦程等人深入探讨了电化学电势对氮掺杂石墨烯(N–C)衬底上SAC中“前沿轨道”的调控。
计算方法
作者使用维也纳从头算模拟软件包(VASP)进行密度泛函理论计算,并使用Perdew–Burke–Ernzerhof(PBE)泛函与投影增强波(PAW)方法来处理核价相互作用,以及将平面波基组的截断能设置为400eV。作者使用p(4×4)的石墨烯超晶胞,并且所有表面都用20Å的真空层隔开。对于倒格空间采样,作者在优化过程中使用5×5×1网格,而在态密度(DOS)计算中使用15×15×1网格。此外,电子步中能量的收敛标准为10–6 eV,而最大力的收敛标准为0.02 eV/Å。
结果与讨论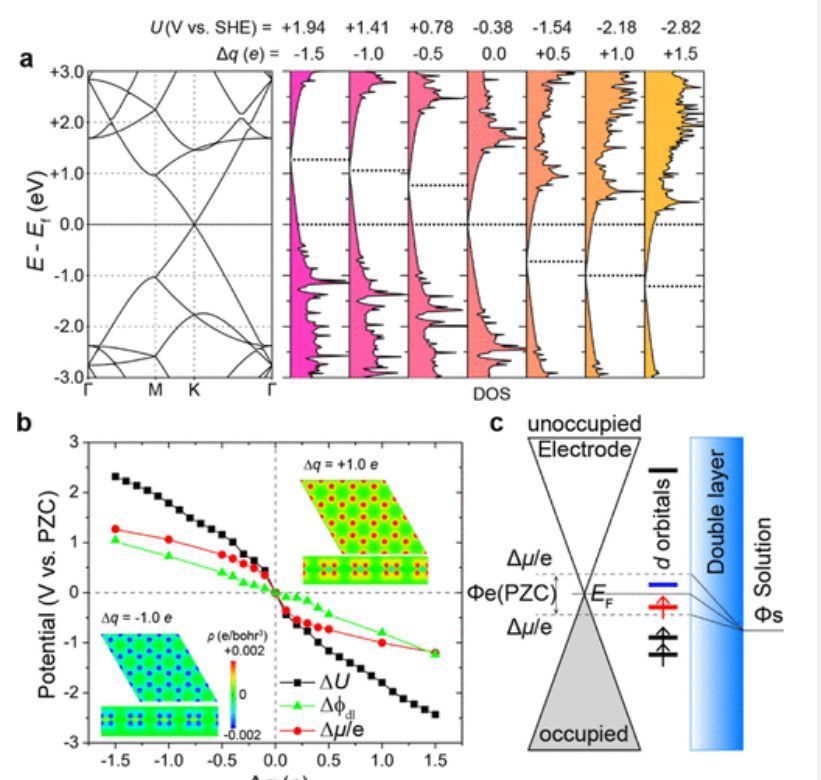
图1 石墨烯的电子性质
如图1a所示,石墨烯的狄拉克点与PZC的K点重合,并且零电荷点(PZC)处的费米能级相对于SHE为-0.38V。作者通过该系统进行电子减法或加法运算,以实现+1.94至-2.82 V的电势范围。如图1b中的黑线所示,PZC附近石墨烯的斜率比其外部区域的斜率更陡,表明PZC周围的电容更低。这一观察结果与狄拉克点的能级偏移一致,如图1b中的红线所示。如果金属轨道的能级位于费米能级的偏移范围内,那么金属活性中心的轨道将被修改。假设中心金属的d轨道刚好位于费米能级之上,增加电子的负电势将导致d轨道的填充,如图1c中蓝色轨道所示。相反,如果中心金属的d轨道刚好低于费米能级,则正电势将从d轨道上移除电子,如图1c中的红色轨道所示。
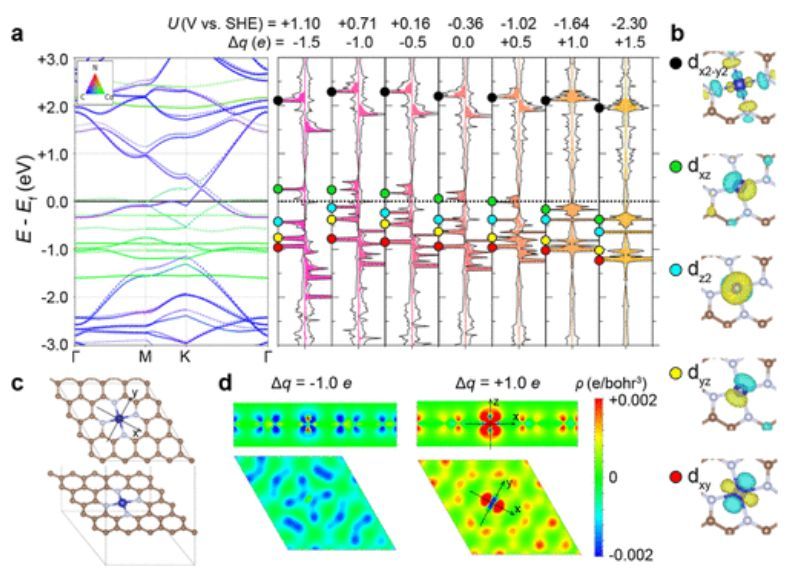
图2 Co/N–C的电子性质
作者以Co/N–C SAC为例来说明中心Co原子的能级变化。如图2所示,PZC处狄拉克点的能级略高于费米能级,Co原子表现出一个未配对dxz电子的自旋极化,PZC位于-0.36 V左右。图2a–c所示,由于其具有沿x方向取向的六元环和沿y方向取向的五元环,向下自旋的dxz轨道能级向上移动到费米能级。dx2–y2轨道明显高于费米能级,不包括其与吸附质的相互作用。鉴于Co具有4s空轨道,Co在PZC的电子结构为d7,氧化态为+2,自旋量子数S为1/2。通过对PZC施加负电势,可以填充未被占用的dxz轨道。通过引入一个额外的电子,电势降至-1.64 V,使Co金属的电子态从d7转变为d8,氧化态为+1,并且没有自旋极化。为了进一步深入了解不同电化学电势下的电子结构,作者比较了PZC和Δq=−1.0e或+1.0e系统的电荷密度差异。Δq在+1.0e时,负电荷密度聚集在Co-dxy轨道上,Δq在−1.0e时,正电荷密度均匀分布在整个Co/N–C系统中(图2d)。
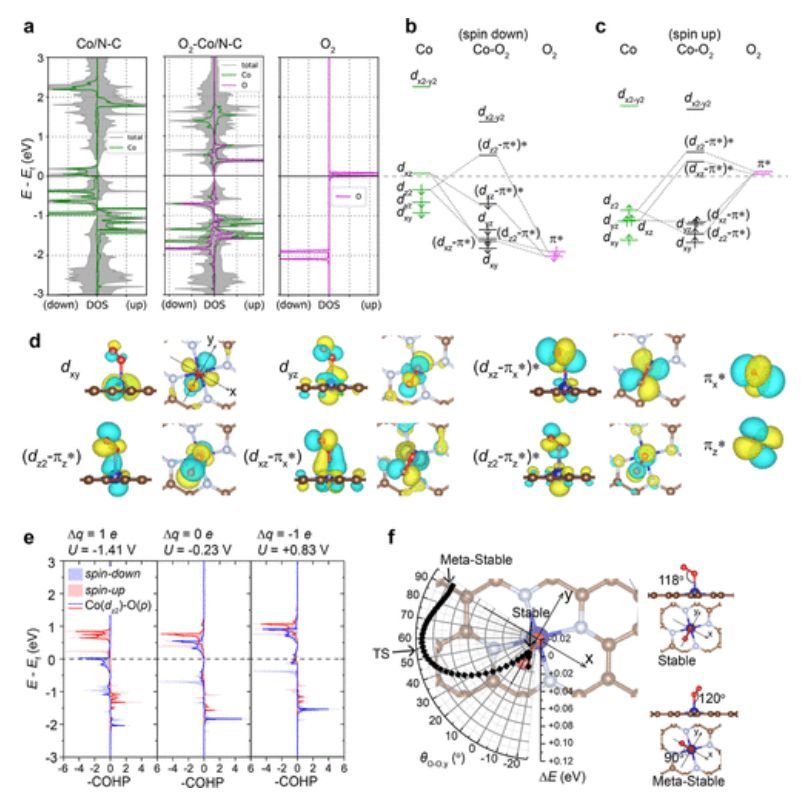
图3 O2在Co/N–C上吸附时的电子性质
如图3所示,对于PZC处的自旋向上相互作用,即O2空的自旋向上轨道与占据的Co-dxz和dz2自旋向上轨道相互作用,这两种相互作用都来自于被占据轨道和未被占据轨道之间的相互作用。形成的被占据dz2–πz*和dxz–πx*轨道的能级下降,而未被占据的(dz2-πz*)*和(dxz-πx*)*轨道的能级上升,从而导致PZC处Co–O对的自旋向上ICOHP贡献为-1.47 eV。另一方面,对于PZC处的自旋向下相互作用,即所占据自旋向下的O2π*轨道与Co原子占据的自旋向下dz2轨道和空dxz轨道相互作用(图3b)。优化后的O–O轴与y轴对齐,满足Co-dxz轨道和πx*轨道之间的轨道对称性。
由于O2吸附后dxz和dz2轨道之间的能量交换,形成的成键dxz–πx*和反键(dxz–πx*)*轨道都位于费米能级以下。来自被占据的dxz–πx*和(dxz–πx*)*轨道的COHP部分相互抵消,因此对Co–O键的ICOHP贡献接近0 eV。此外,由于PZC吸附O2后dxz和dz2轨道之间的能量交换,反键(dz2–πz*)*轨道位于费米能级之上,因此对自旋向下ICOHP的负Co(dz2)–O(p)贡献为-0.51eV(图3e)。
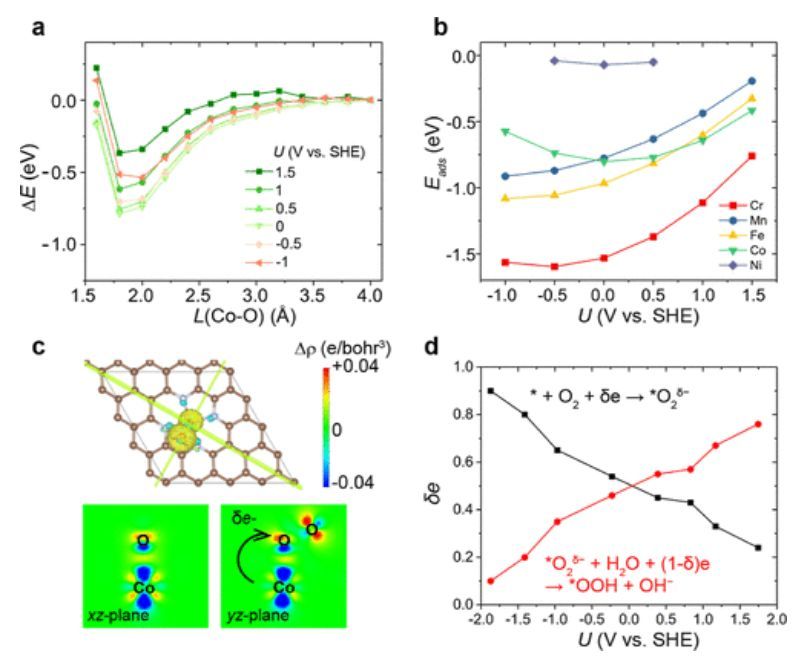
图4 不同电位下M/N–C上的O2吸附
除了Co,作者还探索了在不同电位下,O2在其他SAC上的吸附,包括M/N–C(M=V、Cr、Mn、Fe、Ni和Cu)。如图4所示,除Mn/N–C外,O2在其他3d过渡金属上的吸附能在整个元素周期表中呈现从左到右增加的趋势。具有半填充d5电子结构的Mn2+本质上是稳定的,因此表现出比Fe/N–C和Co/N–C都弱的吸附能。在Ni/N–C和Cu/N–C上,所有dxy、dxz、dyz和dz2轨道都被占据,没有留下合适的空d轨道来稳定半占据的π*轨道。因此,O2在Ni/N–C和Cu/N–C上的吸附能约为0 eV,如图4b所示。
此外,Bader电荷分析显示,从Co/N–C衬底到O2的电荷转移约为0.54 e(图4c)。在ORR电位区域,特别是小于+1.23 V vs SHE的区域,部分ET处的电荷转移(表示为δe)约为~0.4e。同时,部分PCET步骤(从*O2δ−过渡到*OOH)的电荷转移约为~0.6e(图4d)。这表明初始ORR过程是部分ET和部分PCET过程的组合。
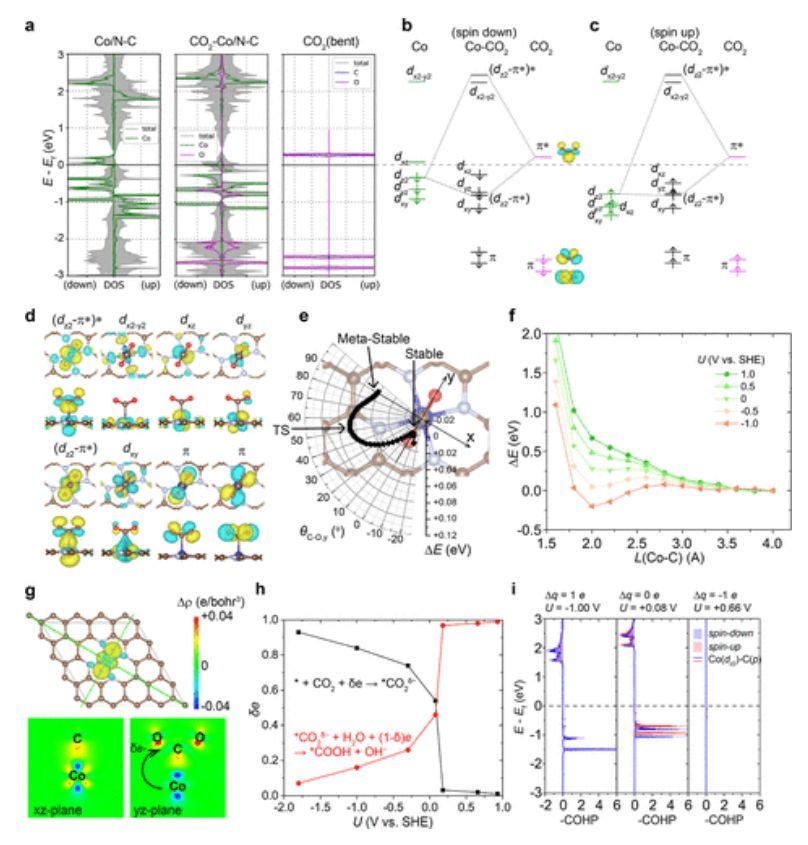
图5 CO2在Co/N–C上吸附时的电子性质
如图5所示,对Co–C相互作用的主要贡献源于dz2和πz*轨道相互作用(图5a–d)。无论是自旋向上还是自旋向下相互作用,dz2和πz*之间的相互作用都会产生一个被占据的成键dz2–πz*轨道和一个未被占据的反键(dz2-πz*)*轨道。因此,在CO2RR的初始ET吸附步骤(*+CO2+δe→ *CO2δ−)中,能量偏移主要由eU项控制,并且在更负的电势下持续下降。
此外,与O2吸附需要与Co-dxz轨道相互作用的情况相反,CO2吸附绕过了与Co-dxz或dyz轨道的直接相互作用。因此,将吸附的CO2分子从y轴旋转到x轴需要克服大约0.07 eV的势垒,最终产生大约0.04 eV的吸热能(图5e)。当电位为正时,CO2在Co/N–C上的吸附表现为物理吸附。只有当电位下降到-0.3 V以下时,吸附才转化为化学吸附,并具有负吸附能(图5f)。因此,在CO2RR条件下,初始步骤在很大程度上为ET步骤,然后是PT步骤,生成*COOH物种(图5g,h)。与O2相比,Co–O对的ICOHP随着U的降低而升高,Co–C对的ICOHP则随着U的减少而降低(图5i)。这表明CO2在Co/N–C上的吸附在更负的电势下变得越来越稳定。
总结展望作者观察到费米能级和d轨道占用随电化学电势的变化而变化,强调了金属的离散原子轨道和基于N–C环境的连续带之间的协同作用。通过使用O2和CO2作为模型吸附质,作者强调了这些变化对吸附能的直接影响,揭示了在负电化学电势下Co/N–C SAC上吸附能的反转。这种见解归因于dxz和dz2轨道的作用,它们对稳定O2的π*轨道至关重要。该工作深入了解了SAC中电子结构和吸附行为之间的相互作用,为电化学过程中增强催化剂设计策略铺平了道路。
文献信息Jin-Cheng Liu et.al Electrochemical Potential-Driven Shift of Frontier Orbitals in M–N–C Single-Atom Catalysts Leading to Inverted Adsorption Energies JACS 2023
ht-tps://doi.org/10.1021/jacs.3c08697


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









