










三元金属化合物因其优异的化学稳定性和催化性能而受到广泛关注。基于此,西北工业大学王俊杰教授(通讯作者)等人结合了机器学习(ML)和高通量计算系统研究了具有P4/mbm空间群的新型A2BC2三元电子化合物。从214个已知A2BC2相的库出发,使用DFT计算了电子局域函数(ELF)的最大值,表明其中42个是潜在的电子化合物。然后在此数据集上训练模型,用于预测通过结构原型产生的14437种假想化合物的电子化合物行为。然后通过高通量计算仔细检验了模型预测的1254个候选电子化合物的稳定性和带电特性。
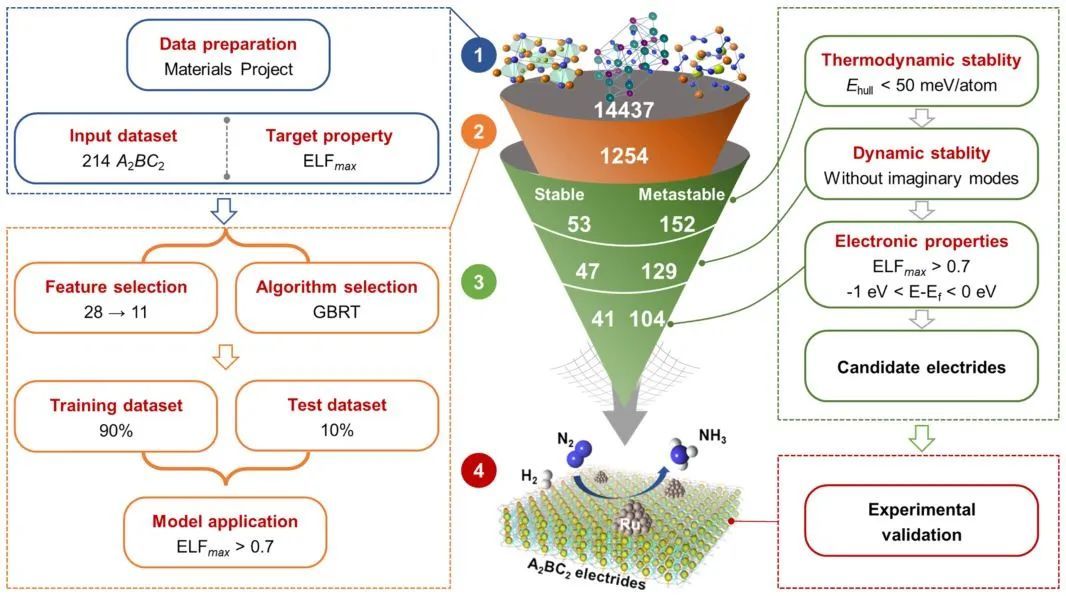
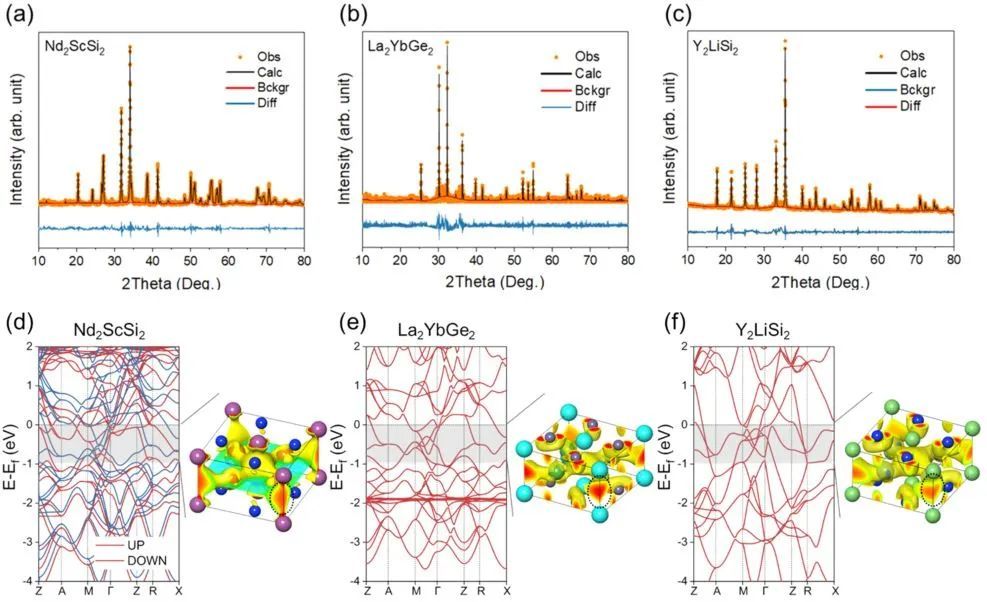
通过这种分层方法,预测了41个稳定的和104个亚稳的新型A2BC2电子化合物。有趣的是,所有三种电子化合物,即缺电子、中性电子和富电子的电子化合物都存在于预测的化合物集中。然后成功地合成了三个最有前途的新型电子化合物(两个富电子的Nd2ScSi2和La2YbGe2,以及一个缺电子的Y2LiSi2),并对其进行了实验表征。此外,当负载Ru时,合成的电子化合物在温和的条件下对NH3合成表现出很高的催化活性。尤其是缺电子的Y2LiSi2,表现出催化活性和化学稳定性的良好平衡,这预示着它在催化中的应用前景。
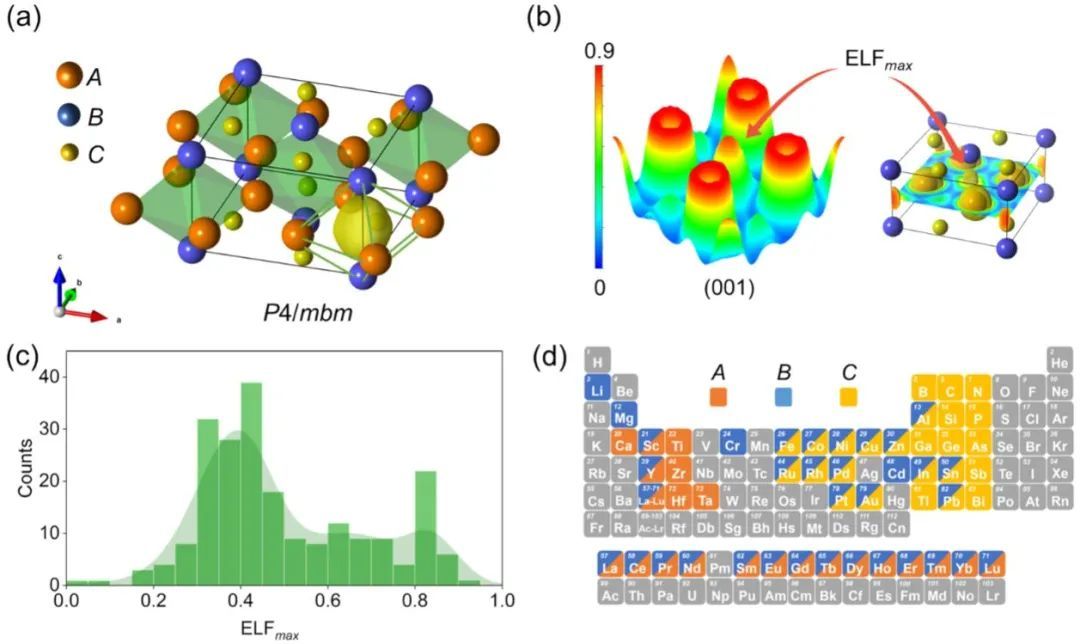
这项研究表明,机器学习可以为带电的理论研究提供有用的见解,同时能够有效地筛选给定结构原型,而不受电荷平衡条件的影响。电子化合物在化学空间中可能并不像人们想象的那样稀有,缺电子的电子化合物不仅是只存在于理论研究中的幻想结构,而且是真实的、潜在有价值的材料。
Machine Learning-Accelerated Discovery of A2BC2 Ternary Electrides with Diverse Anionic Electron Densities. J. Am. Chem. Soc., 2023, DOI: 10.1021/jacs.3c10538.
ht-tps://doi.org/10.1021/jacs.3c10538.


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









