










 本文探讨了电催化研究中过渡金属X化物表面状态的重要性,通过计算和理论分析发现,表面状态对催化剂的活性有显著影响。作者利用表面Pourbaix图和计算方法揭示了X化物在特定电位和pH下的表面覆盖差异,强调了在设计和理解催化机制时考虑表面状态的必要性。
本文探讨了电催化研究中过渡金属X化物表面状态的重要性,通过计算和理论分析发现,表面状态对催化剂的活性有显著影响。作者利用表面Pourbaix图和计算方法揭示了X化物在特定电位和pH下的表面覆盖差异,强调了在设计和理解催化机制时考虑表面状态的必要性。

研究背景
在电催化研究中,由于溶剂与含氢、含氧吸附剂之间存在转换平衡,在特定电化学条件下催化剂的真实表面状态往往被忽略。日本东北大学李昊,悉尼大学魏力与曼彻斯特大学Carmine D’agostino等人通过表面Pourbaix分析,发现许多电催化活性过渡金属X化物(例如,氧化物,氮化物,碳化物和氢氧化物)在pH值和感兴趣的电位下,由于水解离或生成,倾向于具有不同于其原始化学计量形式的表面状态。本文总结了14种条件稳定过渡金属X化物材料的密度泛函理论计算的表面Pourbaix图,发现其中一些表面在中等或高电位下倾向于被含氧吸附剂覆盖,说明通过理论计算(如表面Pourbaix图分析)、原位/操作和反应后实验来分析过渡金属X化物电催化剂的表面状态对于准确理解潜在的催化机制是必不可少的。
计算方法
本文通过VASP量子计算软件包实现催化性质的计算,通过GGA-PBE泛函进行电子交互关联的分析,用投影增强平面波(PAW)方法描述了离子-电子的相互作用,将Kohn-Sham函数在截止动能为400-520 eV的平面波基上展开该函数来处理价电子。本文使用D3方法修正了范德华(vdW)相互作用,采用Nørskov等人的计算氢电极(CHE)方法来计算表面Pourbaix图的,能量作为pH和电势的函数,直到原子间力小于0.05 eV Å−1,所有的原子位置和晶格结构都被完全放松,Z方向真空度设置为12 Å。
结果与讨论
Hansen等人在2008年首次提出的计算不同表面态吉布斯自由能随pH值和操作电位变化的表面Pourbaix图,是分析电化学条件下热力学有利表面态的必要条件。曲面Pourbaix图的计算方法详见补充资料。该方法在分析电催化剂的表面覆盖度方面具有较强的预测能力。图1所示为曲面Pourbaix图的绘制流程图。大量例子都表明,许多不太紧密排列的催化剂,特别是X化物表面,可能与它们的原始形式相比有非常不同的表面状态。不幸的是,无论使用表面Pourbaix分析来揭示表面状态信息是否成功,这种分析方法的重要性通常都被忽视了。
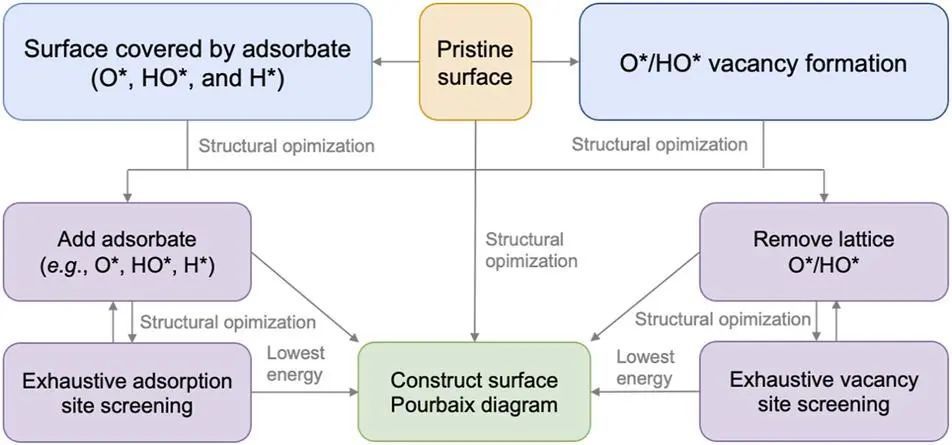
图1 曲面Pourbaix图的绘制流程图
基于上述启发,我们对氮化物催化剂及其衍生物的一些典型表面的电化学状态进行了实例研究,包括TMO(和钙钛矿、尖晶石和金红石),如RuO2、IrO2、RhO2、NiO、β-MnO2、LaMnO3、Sb2WO6、Co2Mo3O8、ZnCr2O4、LaPO4和Ti3C2O2、过渡金属氮化物(ZrN)、过渡金属碳化物(MoC)和过渡金属氢氧化物(NiFe双层状双氢氧化物、LDH)(图2)。所有这些分析都是基于Nørskov等人的工作中开发了氢电极(CHE)方法计算得出的。通过实验和理论分析,证明了这些材料在特定的电催化环境下是稳定的。17,29 - 50,如β-MnO2、LaMnO3、Sb2WO6、Co2Mo3O8、LaPO4和Ti3C2O2、ZrN和NiFe LDH在ORR或OER条件下稳定,而NiO、ZnCr2O4和MoC在相对还原条件下(如HER和NRR)稳定。对于每种材料,选择表面能较低的表面进行分析(表S1-S6)。对于表面Pourbaix图的每个状态,对潜在位点和吸附几何进行了详尽的计算筛选,并丢弃了识别出的高能状态;只画出能量上最有利的状态。由于精确计算不同pH值下的质子化/脱质子化能量学需要充分考虑电场效应,并且受限于用显式模型计算水解离的动力学势垒计算,因此将这些效应集成到表面Pourbaix图计算中仍然是一个悬而未决的问题。在此,本研究在RHE尺度下生成的所有表面Pourbaix图都基于这样一个假设,即许多水解离的动力学障碍可以在感兴趣的电位下被克服。此外,还考虑了溶剂化效应。在IrO2(110)表面上使用显式溶剂化模型的分子动力学模拟结果已被引入,以解释溶剂化效应,HO *和O *的修正值分别为~ - 0.15和~ 0 eV考虑到溶剂化效应主要来源于羟基与水分子之间的氢键作用,不同的TMX材料可能具有相似的溶剂效应。因此,所有与HO *的形成有关的计算(包括HO *的直接生成和O *的质子化)都考虑了一个值的溶剂化修正作为近似值。根据表面Pourbaix图的计算结果,作者进一步研究了在相应的反应条件(比pH值和电位)下,每个反应在其最有利表面上的催化活性。通过计算各关键中间体的吸附能来表征活性。这些中间产物分别为HER的H2O *、OER的O *和HO *、ORR的HO *、CO2RR的COOH *和NNH *。为了进行比较,还计算了上述物种在原始表面的相关吸附能。此外,还引入了标度关系分析,以更深入地了解每种特定反应条件下确定X化物表面状态的重要性。这些表面Pourbaix分析也证明的设计概念的“预催化剂”,许多X化合物的活性相被证明是一个明显被氧化的表面,而不是合成的表面。需要注意的是,不同的表面状态也会导致催化剂的界面性质的变化,如零电荷势和表面疏水性,这将有助于催化机制不同于原始表面。
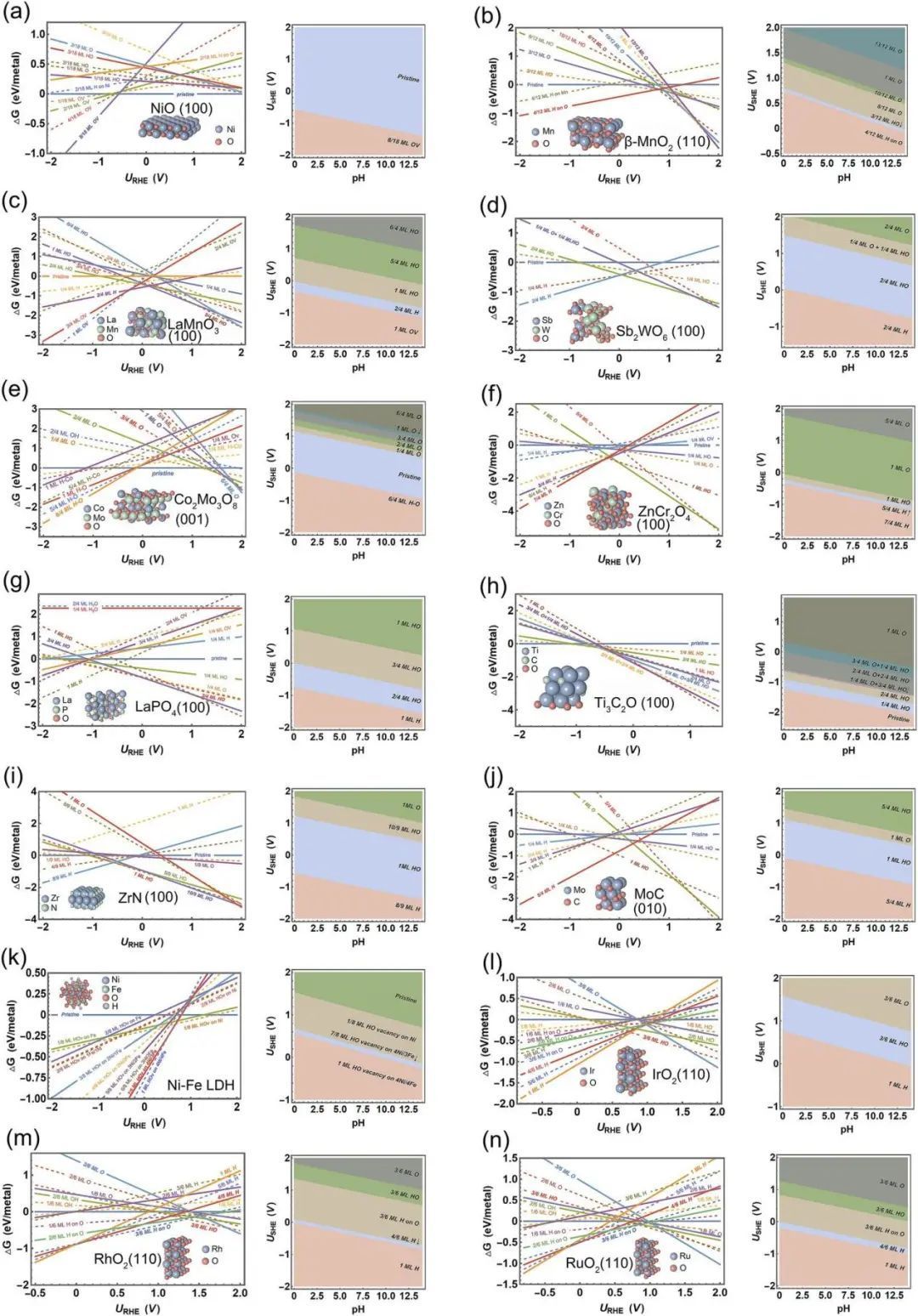
图2 计算了14种典型X化物表面电催化的1D和2D表面Pourbaix图作为pH值和电位的函数。
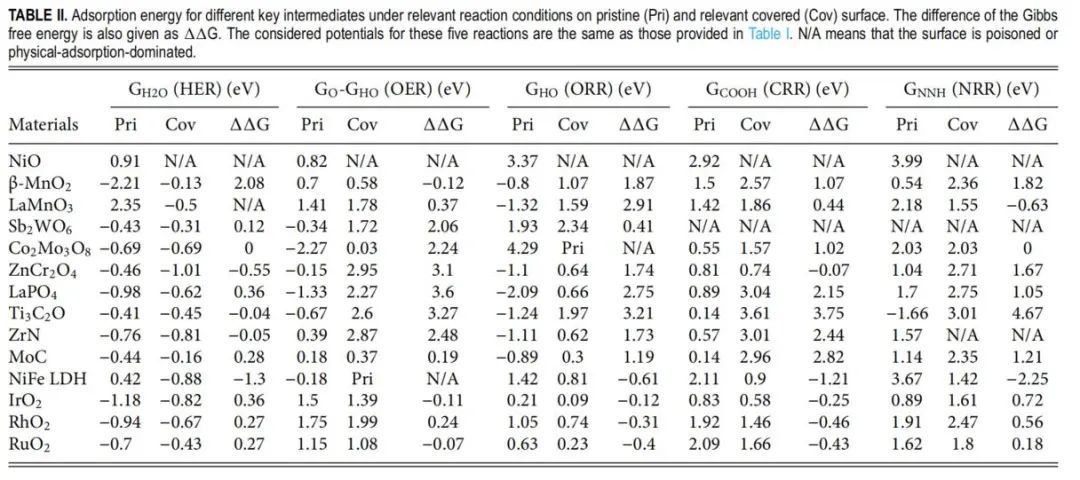
在覆盖前(原始)和覆盖后表面考察了所选14种材料对5种典型电催化的催化活性。结果汇总于表1。在此,选择H2O * (GH2O)、HO * (GHO)、COOH * (GCOOH)和NNH * (GNNH)的吉布斯自由能分别代表HER、ORR、CO2RR和NRR的性能,而O *和HO *之间的吉布斯自由能差用于评估OER活性。作者之所以选择H2O *(而不是H *)来分析HER,是因为大多数TMX在碱性条件下比在酸性条件下更稳定。对于接近1 ML的占用情况,计算了最后覆盖吸附剂的结合能。总的来说,原始表面和被覆盖表面的吸附行为明显不同,这进一步证明了上述发现,也验证了预正表面态研究在电催化研究中的意义,它可以得到完全不同但更现实的结果。需要注意的是,大部分∆G值为正,说明覆盖后吸附变弱。对于少数∆G小于0的情况,这是由于氢键形成产生更强的吸附。
表1 不同关键中间体在相关反应条件下对原始(Pri)和相关覆盖(Cov)表面的吸附能
作者在氧电催化中进行了标度关系分析,为预先进行表面态分析的必要性提供了更深层次的证明。EHO *相对于EO *和EHO *相对于EHOO *的拟合段分别在斜率固定为1.5和1时显示在图3中。在图3(a)中,可以清楚地看到,覆盖后的截距(ORR/OER电位下)与TMX原始表面(蓝线)相比发生了显著变化,表明在中等或高电位下O *键合强度较弱。即与自由基分子(例如,HO *和HOO *)相比,O *的吸附对表面覆盖度更敏感,更“拥挤”的表面会导致对O *更强的排斥相互作用,从而削弱其成键。这可能导致RDS(速率决定步骤)的显著切换以及ORR和OER的最佳反应活性的差异。对于EHOO∗和EHO∗的标度关系,截距几乎是通用的。在确定和分析过渡金属化合物催化剂的活性位点之前,应确定反应条件下的表面状态(如pH值和电位),如果忽视了这一点,将会误导结果。最后但并非最不重要的是,由于这项工作的重点是计算Pourbaix图,作者已经做了一些近似,如修正pH值和溶剂化效应如上所述。鉴于以上研究结果显示了初步确定表面状态的重要性,未来的工作将集中在综合pH、电和溶剂化效应的全面和精确计算上,以深入了解这一问题。
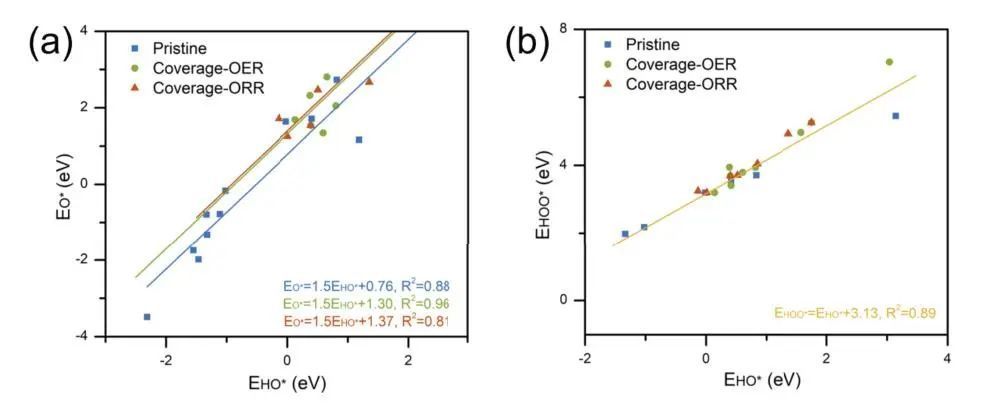
图3 氧电催化的标度关系
结论展望
当分析过渡金属化合物作为电催化剂时,表面状态不应该被跳过——排除这一重要的特性可能会导致错误的反应机制和活性位点的分配。除了理论表面Pourbaix图分析外,也可以通过原位和反应后光谱(如红外、拉曼、x射线光电子和x射线吸收光谱)和显微技术(如透射镜显微镜和原子力显微镜)耦合分析,为描述表面性质提供实验信息。
文献信息
Liu, H., Jia, X., Cao, A., Wei, L., D'agostino, C., & Li, H. (2023). The surface states of transition metal X-ides under electrocatalytic conditions. The Journal of Chemical Physics.
ht-tps://doi.org/10.1063/5.0147123


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









