







 作者信息
作者信息
第一作者:Zan Lian 通讯作者:Núria López
通讯单位:加泰罗尼亚化学研究所
成果速览
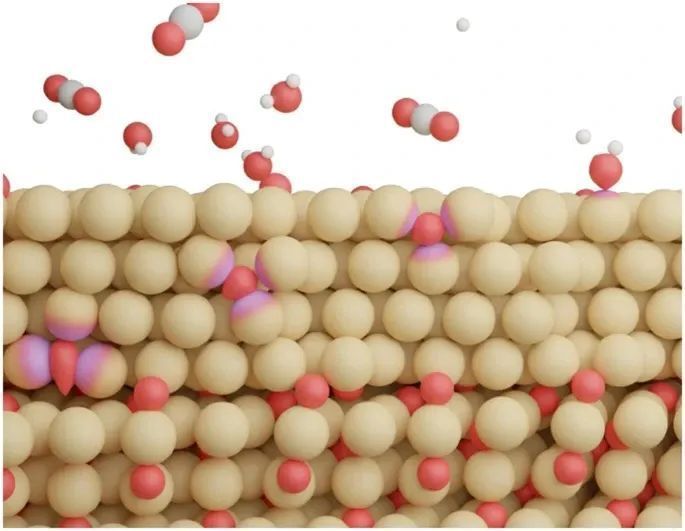
本研究通过大规模分子动力学模拟结合精确的神经网络势能,探讨了氧化物衍生铜(OD-Cu)在电化学CO2还原反应中的氧浓度稳定性与寿命。
研究发现,OD-Cu的氧浓度与pH值、电位和比表面积密切相关。在长时电化学实验中,OD-Cu可完全还原为铜,但去除所有捕获的氧需较长时间。该研究为理解OD-Cu催化剂的演变和活性位点的本质提供了深刻见解。
图文导读
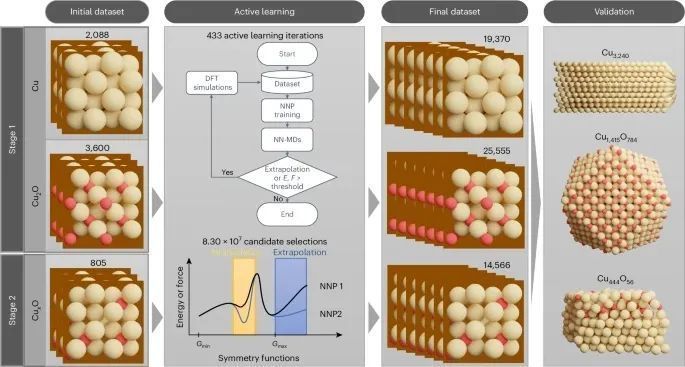
图1:展示了构建神经网络势能的计算模拟方法,包括数据收集的两个阶段和通过主动学习过程进行的迭代。
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1377
1377

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









