










研究背景
由石墨烯上两个相邻的单原子位点组成的双位点催化剂表现出了良好的电化学氧/析氢反应(OER/HER)催化活性。然而,OER/HER在双位点催化剂上的电化学机理仍不明确。在本研究中,松山湖材料实验室研究团队采用密度泛函理论计算研究了O−O/HER对双位催化剂的催化活性。具体来说,这些元素步骤应该分为两类:一类是需要由电极电位驱动的质子耦合电子转移的进化步骤(PCET步骤),另一类是在温和条件下自然发生的没有PCET的步骤(非PCET步骤),这将对指导合理设计电化学反应的有效双位点催化剂发挥关键作用。
计算方法
本文采用Vasp软件包对其催化性能及电子性质进行理论研究,计算基于广义梯度近似(GGA)下的PBE泛函计算电子交换相关性,离子-电子相互作用采用PAW方法进行表征。应用Grimme的DFT-D2方法描述了金属氧化物与7个掺杂氢原子之间的范德华(vdW)相互作用,截断能设置为500 eV,将最大力的收敛公差设为0.02 eV·Å-1,能量收敛公差在1×10-5 eV。利用推动弹带法(NEB)对10个势能面上的一阶鞍点进行了定位,并通过后续的振动频率计算进行验证。
结果与讨论
CHE模型是一种被广泛使用的方法,可以有效地筛选OER/HER催化剂,并寻找最佳的催化剂。对于OER,用计算出的过电位(η)来表示催化活性。O−O直接耦合形成O2的步骤与质子耦合电子转移(PCET),因此,这是一个与电位无关的过程(图1a,b)。实际上,已经报道了一种计算双位点η的方法,这表明η很可能是由无关的非PCET步骤贡献的。然而,使用这种方法来计算η来评估最近报道的双位点催化剂的OER活性效果不佳。其中,O−O在双位点上的直接偶联机制的η(1.17V),甚至高于单位点上的亲水机制(1.14 V),说明双位点的活性弱于单位点。但这样的理论结果与实验结果完全相反。在这方面,作者认为,过电位的单个参数不足以评价具有O−O直接偶联机制的双位点催化剂的OER活性。对于具有O−O (H−H)直接耦合机制的OER (HER),单元步长可分为两类:PCET步骤和非PCET步骤(图1b)。与势相关的η应由势相关的PCET步骤贡献,而与势无关的非PCET步骤贡献。O−O(H−H)直接耦合的独立非PCET步骤的活化势垒(Ea)是另一个与化学反应速率密切相关的重要因素。在HER与H−H直接耦合机制中也发现了类似的矛盾关系。因此,在评价OER(HER)与O−O(H−H)直接耦合机制的催化活性时,应同时考虑过电位(由电位依赖的PCET步骤贡献)和活性势垒(来自与电位无关的非PCET步骤)。
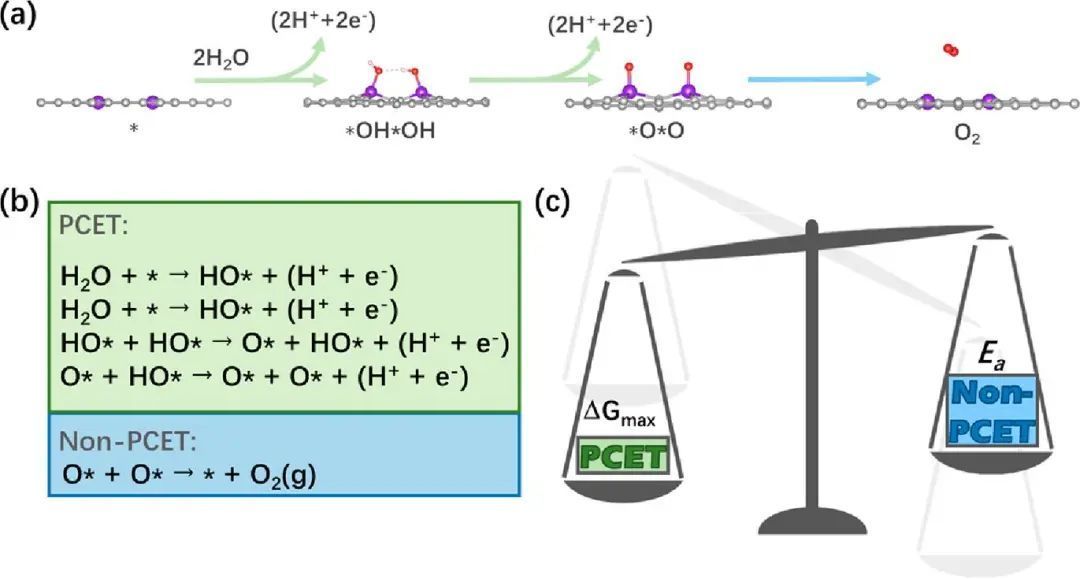
图1 反应机理说明
在石墨烯上由两个相邻的单原子位点组成的双位催化剂用于催化化学反应。MD计算表明,所研究的双位点催化剂是稳定的。研究OER机理的亲水机制和O−O直接耦合机理。亲水机制包括四步PCET过程和*OH、*OOH和*O的三个中间体(图2a)。亲水机制的决速步是形成Mn/Fe/共偶位(OOOH和镍二位(OER)的OH形成步骤。Ni双位(OER)的最低过电位为0.87 V。可以看出,所有这些中间体都以相同的吸附构型结合在单位点上:通过单键氧连接,这表明它们不能在不影响另一个单键氧的情况下独立调整,从而导致彼此之间的线性尺度关系。如图2b、c所示,ΔG*OH与ΔG*O(或ΔG*OOH)之间的比例关系与之前的报道一致,基本上形成了火山曲线规律。
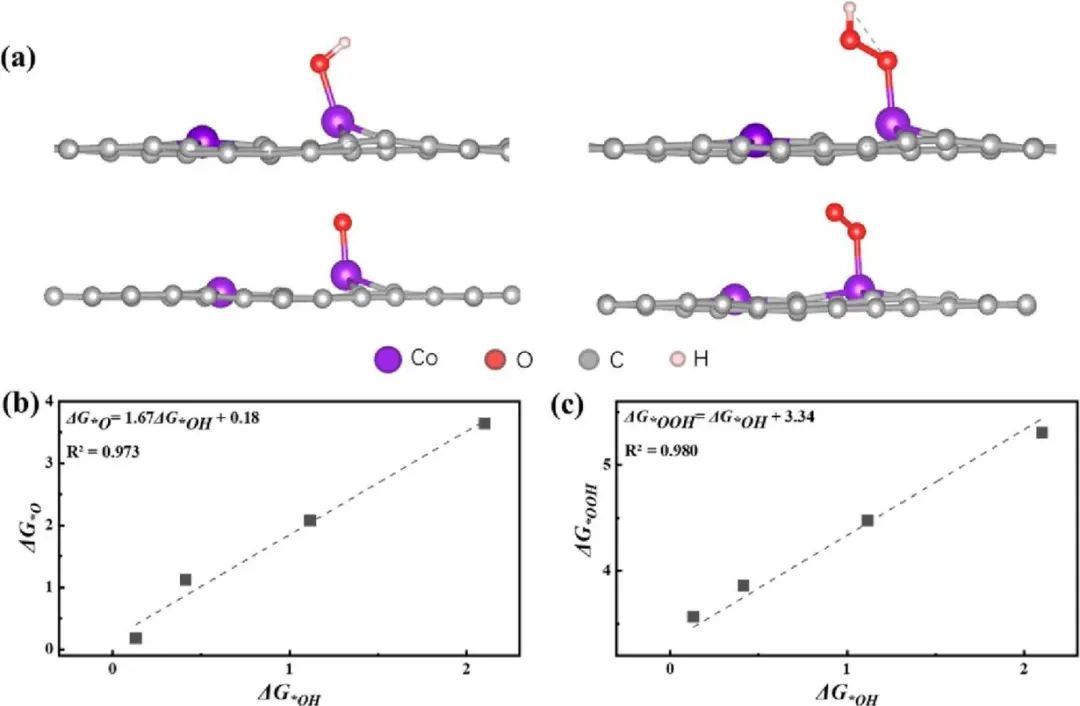
图2 OER反应结构模型及特征标度关系
以共双位点(OER)为例,利用O−O直接耦合机制构建了OER的自由能分布图(图3b)。可以看出,PCET步长的自由能变化分别为0.98(ΔG1)、1.00(ΔG2)、1.23(ΔG3)和1.16(ΔG4),表明PDS是*OH−*OH脱氢生成*OH-*O,与ΔG3相结合,形成ΔGMax(1.23 eV)。对于O−O直接耦合的末端非PCET步骤,它是一个热力学吸热的。非PCET步骤可以在温和的条件下发生,O−O直接耦合形成吸附的O2的活性能(Ea)。Ea为0.73 eV,表示O−O直接耦合过程在较温和的条件下进行。同时,O2解吸的活性势垒为0.65eV,表明所形成的O2的解吸是可行的。因此,共双位(OER)可以用于OER,相应的过电位为0V,这意味着共双位(OER)可以作为理想的OER催化剂,而不消耗额外的能量。Ni -双位点(OER)的自由能变化分别为2.10 (ΔG1)、2.13 (ΔG2)、1.67 (ΔG3)和1.49 eV (ΔG4),表明PDS是一个形成*OH-*OH的步骤,ΔGMax为2.13 eV。同时,O−O耦合的自由能变化为放热(ΔG5 =−2.47 eV),这意味着O-O耦合形成O2分子是容易发生的。从图3c可以看出,ΔG5和E*O呈线性函数关系,ΔG5和Ea之间呈正相关关系,可以看出Ea的比值普遍大于ΔG5。因此,Ea的值是评估非PECT O−O直接耦合可行性时的参考值。图3d所示,过电位随着TM金属丰度的增加而增大。然而,Ea随着TM金属丰度的增加而减小。换句话说,E*O较弱的结合能通常与较高的过电位和较低的活性势垒有关。
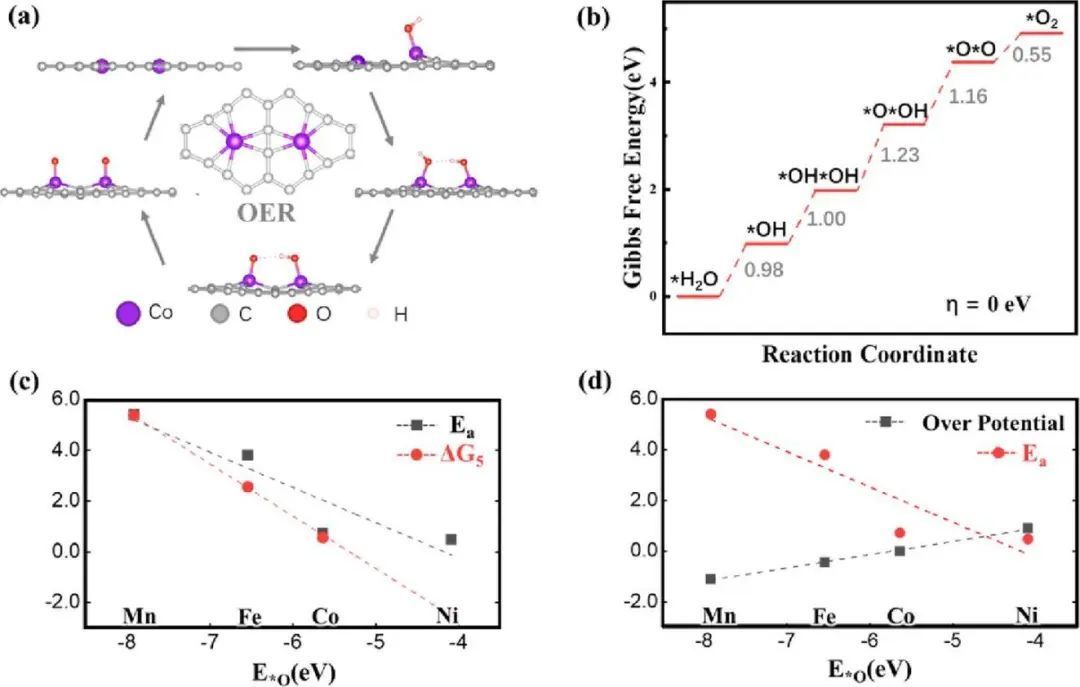
图3 催化剂的催化能力
双位点催化剂也用于HER,研究的双位点如图4a所示,TM = Mn/Fe/Co/Ni)。MD计算表明。作为HER活性的可靠描述符,我们对单个位点上的|ΔG*H|进行了检测。HER活性随着TM金属丰度的增加而增加,而Mn-双位点(HER)的单位点具有最好的HER活性。在双位点上,很可能进行H−H直接偶联形成氢分子。如图4b所示,HER的自由能分布表明,ΔG1的自由能垒需要通过电极电位克服自由能垒(0.82 eV)。对于H−H直接耦合的末端非PCET步骤,它是热力学吸热的。为了检验H−H直接耦合是否能在温和的条件下发生,计算H−H直接耦合形成吸附的H2的活度能(Ea)。Ea为0.68 eV,表示H−H直接耦合过程发生在温和的条件下。基本上,数值关系(ΔG1 +ΔG2 =−ΔG3)表示PCET的自由能变化总量与非PCET阶的自由能变化呈负相关关系。具体来说,PCET的ΔGMax随着TM金属丰度的增加而增加。然而,非PCET的Ea随着TM金属丰度的增加而减小。

图4 催化剂HER活性
结论与展望
综上所述,我们采用DFT方法和CHE模型研究了O−O/H−H直接偶联机理在双位点催化剂上的OER/HER,数值关系(ΔG1 +ΔG2 =−ΔG3)表示PCET的自由能变化总量与非PCET阶的自由能变化呈负相关关系。这些发现有助于对双位催化剂上的OER/HER的深入了解,并促进催化反应双位催化剂的研发。
文献信息
Hu, M., Ye, K., Zhang, G., Li, X., & Jiang, J. (2023). Insight into the Mechanism for Catalytic Activity of the Oxygen/Hydrogen Evolution Reaction on a Dual-Site Catalyst. The Journal of Physical Chemistry Letters, 14, 2201-2207.
ht=tps://doi.org/10.1021/acs.jpclett.3c00168















 2万+
2万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









