











成果简介
无序固溶体合金作为一种潜在的催化剂,可以提供丰富的催化位点。然而,从每个单独的位点准确评估它们的活性结构和整体活性仍然是一个艰巨的挑战。
中国科学技术大学宋礼教授、武晓君教授等人采用基于密度泛函理论和机器学习的方法,获得了Pt-Ru合金作为析氢反应模型多位点催化剂的大量位点。随后,采用一系列统计方法揭示几何结构与整体活性之间的关系。根据金属元素的径向频率分布和ΔGH的分布,作者确定了Pt和Ru分别占据表面和次表面是最佳的结构。特别是,等价位点比的概念预测了Ru含量为20~30%时的总活性最高。此外,还合成了一系列Pt-Ru合金来验证所提出的理论。这为理解合金催化活性的起源提供了重要的见解,从而将更好地指导靶向多位点催化剂的合理开发。
相关工作以《Analyzing the Active Site and Predicting the Overall Activity of Alloy Catalysts》为题在《Journal of the American Chemical Society》上发表论文。该论文得到了科技部重点研发计划(2022YFA1504104,2022YFA1605400)等项目的支持。
图文导读
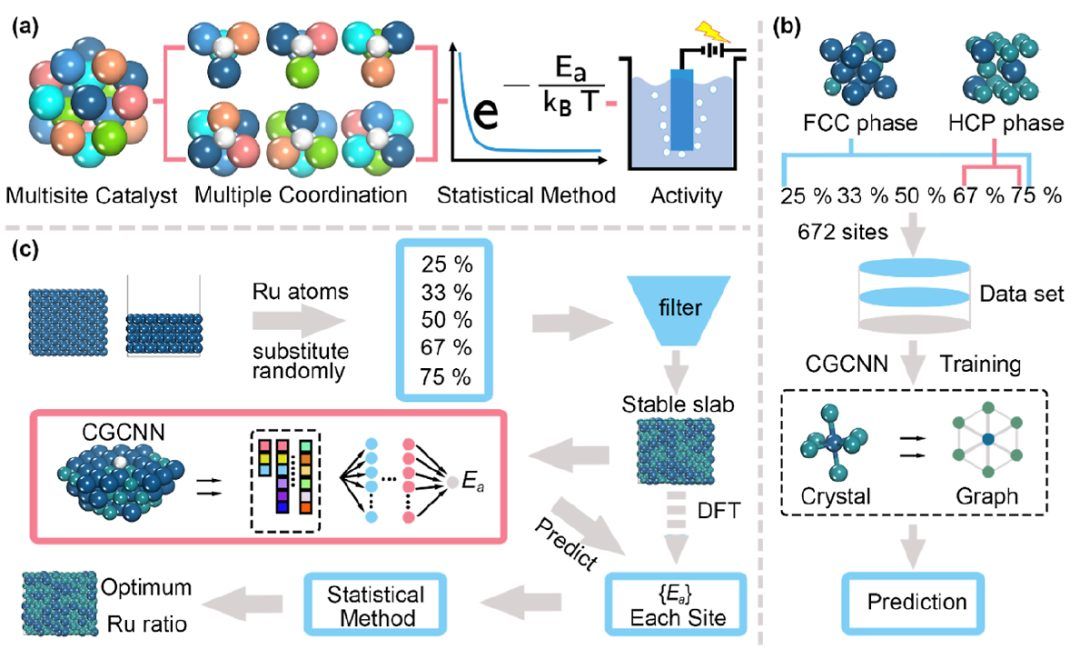
图1 概念框架示意图及计算流程
图1说明了本研究的目的和程序框架。作者采用统计方法阐明了活性局部结构(ALS)与Pt-Ru多位点催化剂的内在活性之间的关系,并构建了整体活性模型(图1a)。利用DFT数据训练的机器学习(ML)模型来加快计算过程(图1b)。通过随机替代生成的多种Pt-Ru无序固溶体(DSS)合金催化剂,筛选稳定表面并用作ML模型的输入。然后利用结构数据和预测的吸附自由能数据进行后续的总体活性分析和计算(图1c),以寻找最佳的合金。
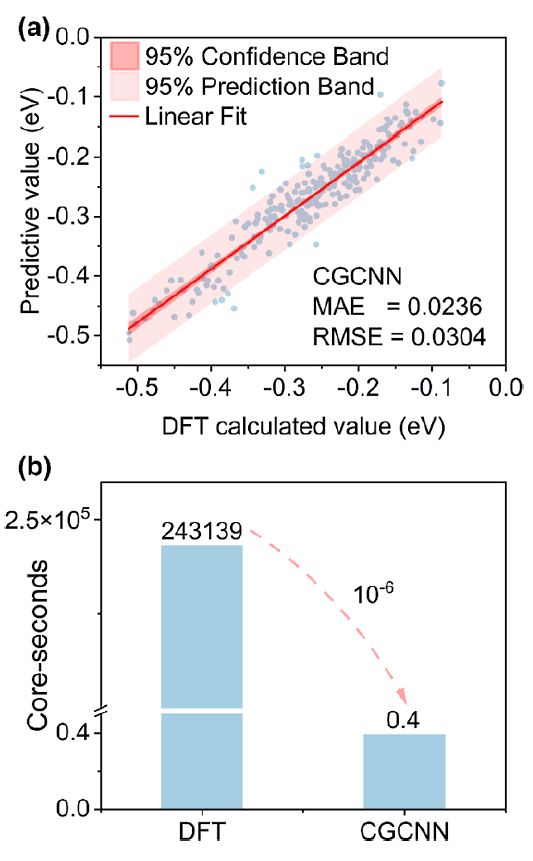
图2 机器学习测试结果 为了克服合金的随机性带来的挑战,采用DFT数据训练的CGCNN模型来计算H吸附的自由能。模型的准确性将使用平均绝对误差(MAE)和均方根误差度量来评估。如图2a所示,与Dimenet++(0.0244 eV)、Schnet(0.0259 eV)和GATGNN(0.0273 eV)相比,CGCNN在测试集上的预测结果(MAE:0.0236 eV)具有更高的准确性。使用CGCNN来预测所有位点的ΔGH。通过规避计算密集型的Kohn-Sham方程的求解,与DFT方法相比,CGCNN减少了6个数量级的计算资源(图2b)。
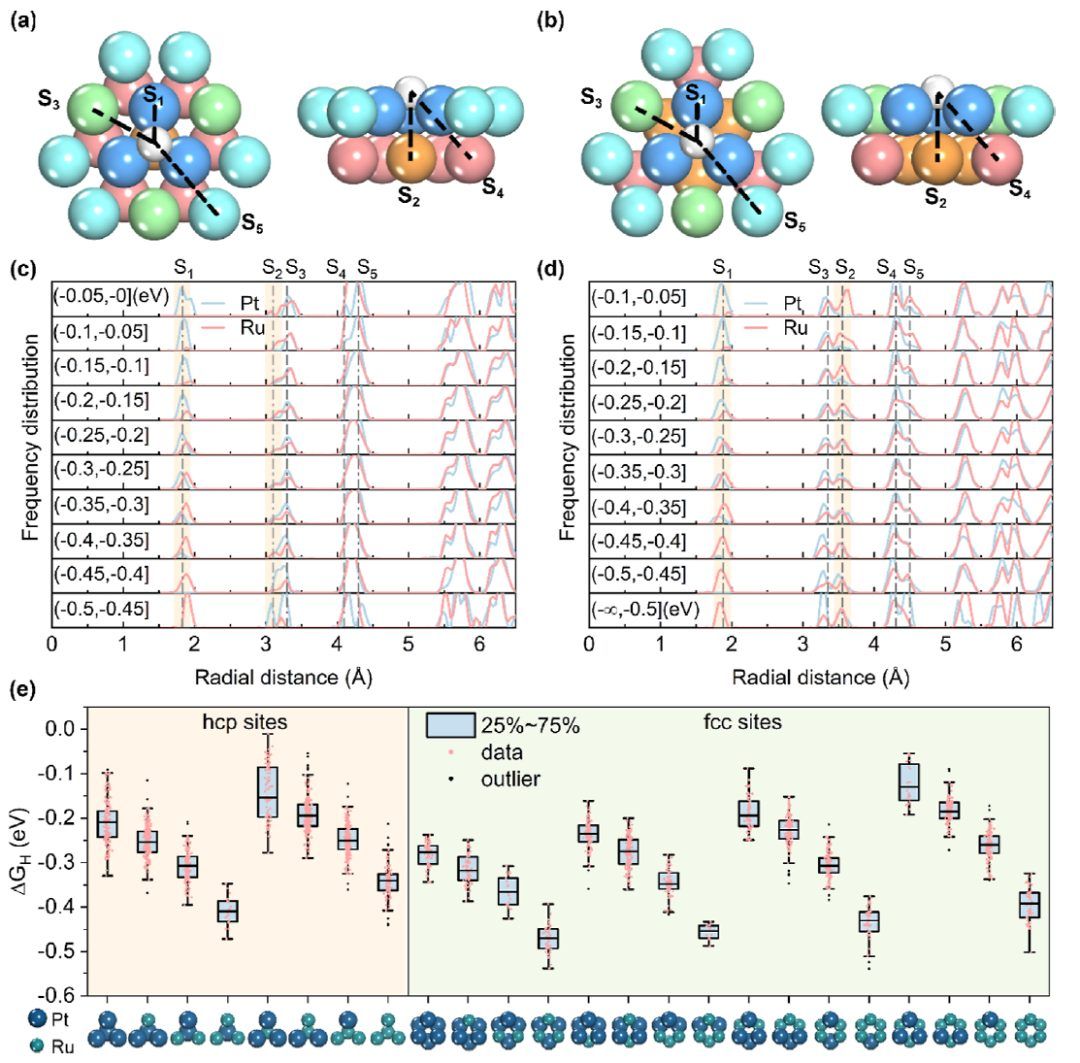
图3 配位环境的统计分析
壳层的目的是描述站点的局部配位环境。与氢原子距离相近的金属原子聚集在同一壳层中。hcp和fcc位点都有5个壳层(图3a、b中标记为S1、S2、S3、S4和S5)。由于吸附质分子之间的距离和静电屏蔽效应显著,S5壳层以外原子的影响可以忽略不计。Sabatier原理解释了最佳表面,它具有中等的结合能(ΔGH≈0 eV)。因此,根据不同ΔGH间隔的平均径向频率分布,对ALS进行统计和分析。 图3c,d显示了元素的径向频率分布,代表了hcp和fcc位点的壳结构。这种分布是通过分析结构数据中靠近H原子的金属元素的位置和频率得到的。与其他壳层相比,Pt和Ru元素在S1和S2壳层内的分布在不同的吸附自由能区间上表现出显著的变化。当吸附自由能接近于零时,hcp和fcc位点的S1壳层以Pt为主,S2壳层以Ru为主。在强吸附位点,元素的分布呈现相反的趋势。从强吸附到弱吸附,各元素分布均匀。基于上述分析,假设吸附自由能与S1和S2壳层中元素的分布有很强的相关性。此外,通过随机改变S3到S5配位壳层中的单个原子进行了测试。原子的修改对ΔGH的影响很小。 因此,元素在核-壳中的排列可以作为区分位点类型的一种手段。将2400个站点结构分成不同的分类将有助于以后的统计分析。增加壳层的数量会增加分析的复杂性,同时降低位点类型的代表性。因此,在随后的讨论中,根据S1和S2壳层中Pt和Ru元素的组成来区分位点类型(S1|S2,hcp:PtxRu3-x|PtyRu1-y,fcc:PtxRu3-x|PtyRu3-y)。
为了进一步探索结构-活性关系,分析了ΔGH在所有类型位点上的分布。图3e表明,每种位点类型的ΔGH都是一个分布。这主要是由于S3、S4和S5的轻微影响。尽管如此,ΔGH的分布与位点的类型之间存在明显的相关性。hcp和fcc位点的参考点分别为Pt3|Pt1和Pt3|Pt3,它们与纯Pt的组成接近。增加S1壳层中Ru原子的比例可以增强吸附能力,而S2壳层中Ru原子的存在则相反,导致吸附能力减弱(例如,对于H的吸附强度,Pt3|Pt3<Pt2Ru1|Pt3<Pt1Ru2|Pt3<Ru3|Pt3;而Pt3|Pt3>Pt3|Pt2Ru1>Pt3|Pt1Ru2>Pt3|Ru3)。这一现象在hcp和fcc位点均有发现。
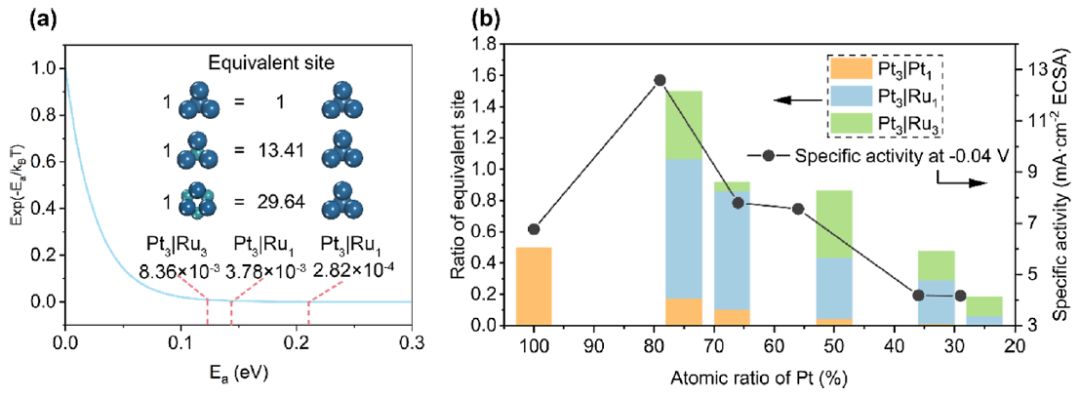
图4 多位点合金催化剂模型的验证
将纯Pt计算数据直接纳入与Pt-Ru合金比较的多位点催化剂模型可能会导致相当大的不准确性。为了解决这个问题,使用Pt3|Pt1来表示纯Pt的位点,因为纯Pt的hcp位点可以被视为这类位点的特殊实例。选择Pt3|Pt1位点作为参考,评价其他位点的活性。根据多位点催化剂模型,位点的重量与其活化能呈指数关系(图4a)。等效位点的个数可以通过任意位点与参考位点的相对权重得到。例如,Pt3|Ru1和Pt3|Ru3分别是hcp和fcc位点中活性最高的位点,其活性相当于Pt3|Pt1位点的13.41和29.64倍。
图4b显示了5种Pt-Ru合金的ESR。随着少量Ru的加入,ESR迅速增加,并在Pt含量达到75%时达到峰值。随着Ru浓度的增加,ESR逐渐降低。在低浓度的Ru下,Pt-Ru合金催化剂的活性高于纯Pt催化剂,原因有二。首先,正如前面所讨论的,Ru原子倾向于远离表面,这有利于形成高活性的Pt3|Ru1和Pt3|Ru3位点。第二,现阶段催化剂中仍有相当数量的Pt3|Pt1位点。Ru的进一步增加将抑制这两个积极因素。 最后,对Pt-Ru合金催化剂样品进行了电化学实验,以确定多位点催化剂模型得出的结果是否与实际相符。在电流密度为10 mA cm-2时,Pt79Ru21合金催化剂的过电位为17 mV,而Tafel斜率为18 mV dec-1。此外,Pt79Ru21在18 h的计时电位测试中显示:电位仅下降8 mV,表明其活性位点结构稳定。实验结果表明,比活性在Ru含量为21%时达到峰值,然后随着Ru含量的增加逐渐降低,这与ESR一致(图4b)。
文献信息
Analyzing the Active Site and Predicting the Overall Activity of Alloy Catalysts,Journal of the American Chemical Society,2024.















 1419
1419

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









