








单细胞RNA测序(scRNA-seq)识别组织内的细胞亚群,但不能捕获它们的空间分布,也不能揭示细胞间通讯的局部网络。空间转录组能够对细胞进行定位,却无法准确地在组织中产生深层单细胞分辨率的转录组图谱。成功整合单细胞和空间转录组数据的分析将有助于理解细胞类型分布的结构以及构成这种结构的细胞间通讯的假定机制。
2021年,美国斯坦福大学回顾了整合scRNA-seq与空间转录组数据的多元化计算方法【1】。整合策略主要分为2种方式,去卷积(Deconvolution)以及映射(Mapping)。
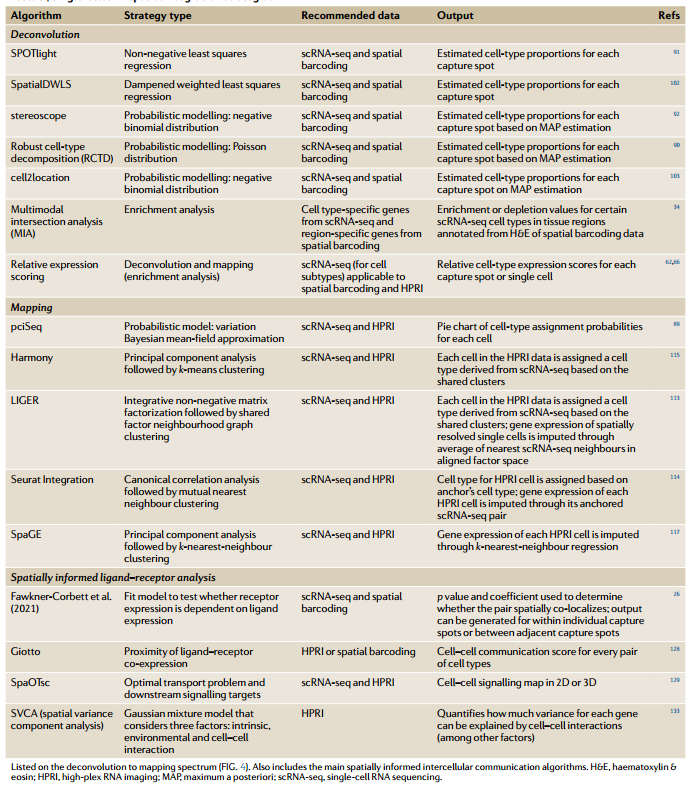
图1 单细胞和空间整合策略
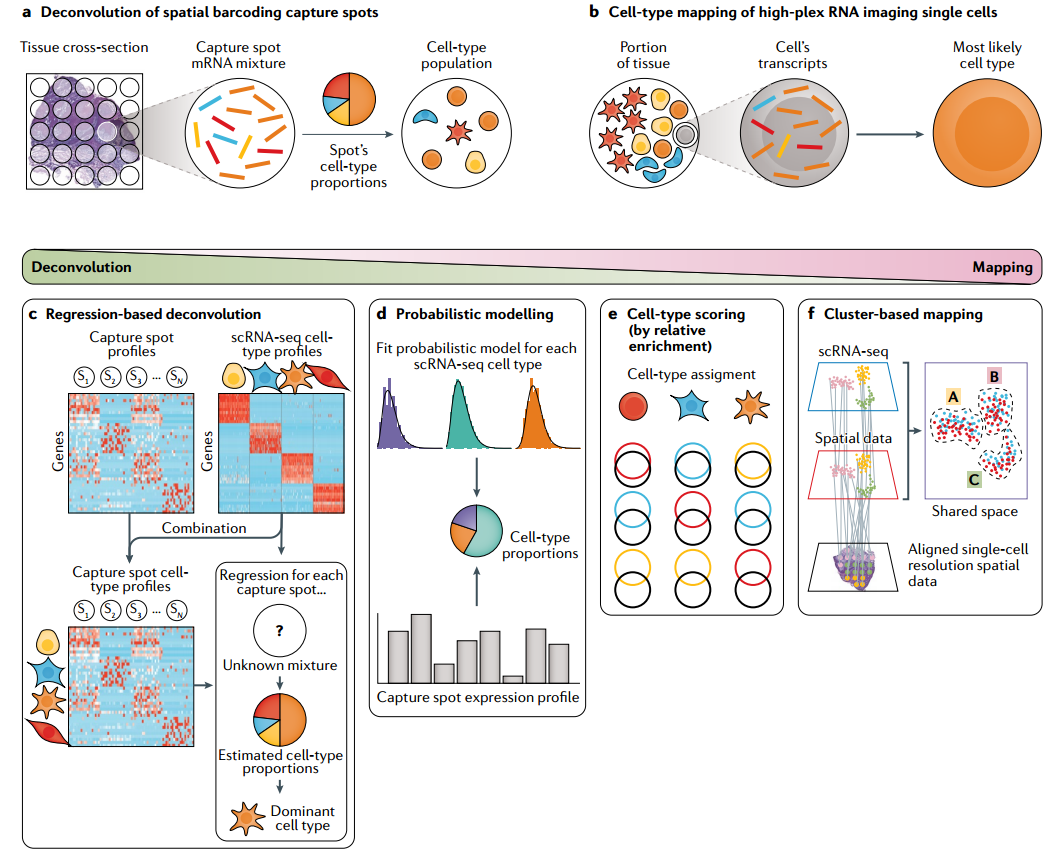
图2 反卷积和映射方法
一、利用scRNA去卷积(Deconvolution)
ScRNA deconvolution通过对实验过程建模来重建空间转录组观测结果,并估计每个孔中的细胞类型组成。这种方法通常采用非负矩阵分解(NMF)或线性模型,旨在根据单细胞数据,从每个捕获点的mRNA转录物的混合物中分离出离散的细胞亚群。代表性的工具有SPOTlight、CARD、RCTD、Cell2location等。
01 SPOTlight
SPOTlight【2】全称为Spatially complete stranscriptomics light,是专门为10x Visium空间转录组学技术开发的。在空间转录组技术中,由于技术限制,单个spot往往包含多个细胞类型的混合信号。SPOTlight可以结合单细胞转录组RNA测序信息反卷积deconvolute空间数据,识别每个spot中的细胞类型和比例。
-
【文献案例一】

-
期刊:Clinical and Translational Medicine
-
影响因子:7.9
-
单细胞技术:10x scRNA-seq;10x空间转录组
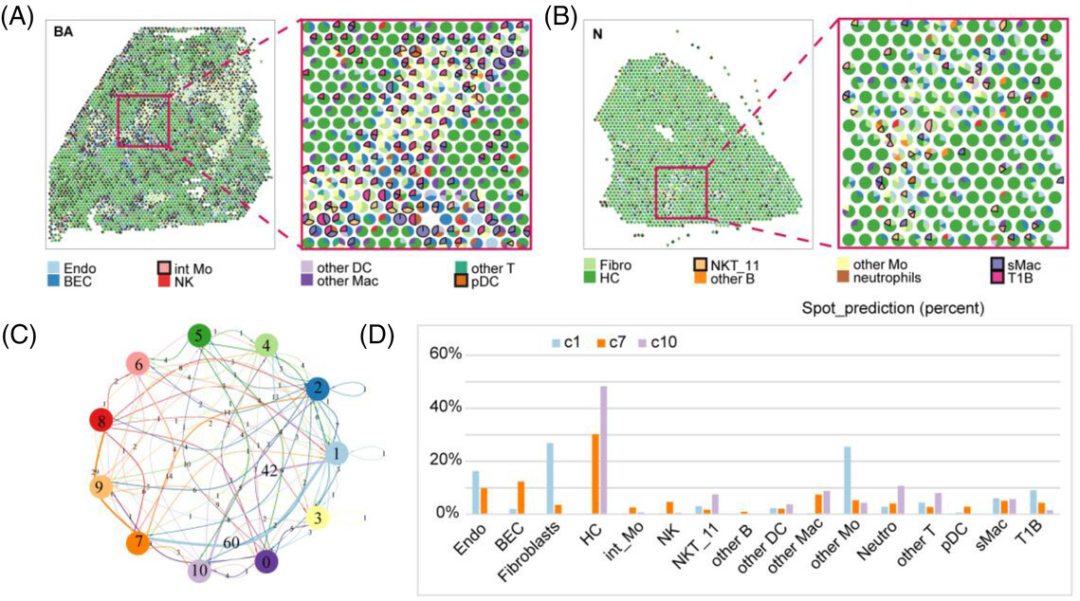
胆道闭锁(BA)是一种破坏性的炎症闭塞性新生儿胆道疾病。它破坏肝外和肝内胆管,扰乱胆汁流动并迅速产生严重的肝纤维化。本文结合scRNA-seq和空间转录组学两种新的研究方法,揭示BA肝纤维化生态位的细胞、分子和空间免疫微环境,旨在表征目前未知的细胞类型、细胞间相互作用和关键分子。文章利用SPOTlight对特定纤维化相关免疫细胞进行了定位。
02 CARD
CARD【3】(conditional autoregressive deconvolution)是一种基于条件自回归(conditional autoregressive;CAR),并结合单细胞测序数据中细胞类型特异性表达的信息进行反卷积分析的方法。基于细胞类型组成中的邻域相似性,CARD可以进一步用于构建细胞类型组成和基因表达水平的精细空间图。
-
【文献案例一】

-
期刊:Journal of Genetics and Genomics
-
影响因子:6.6
-
单细胞技术:华大C4;stereo-seq
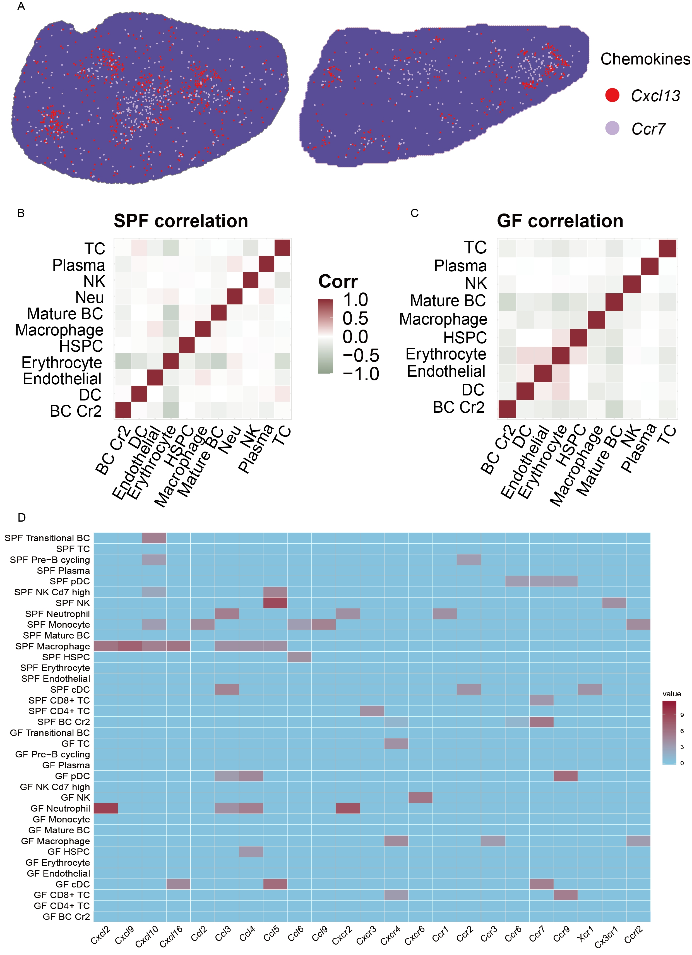
肠道微生物与宿主表现出复杂的相互作用,并在整个生命周期中塑造生物体的免疫系统。脾作为最大的次级淋巴器官,具有广泛的免疫功能。为了探索微生物群在调节和塑造脾脏中的作用,本文采用基于无菌(GF)小鼠的scRNA-seq和Stereo-seq技术检测了脾脏组织大小、解剖结构、细胞类型、功能和空间分子特征的差异。文章使用CARD对小鼠的空间细胞类型进行了共定位。
03 RCTD
RCTD【4】(Robust cell-type decomposition),是一种空间反卷积工具,整合了统计学模型,能够利用带注释的scRNA-seq数据来定义空间转录组学数据中空间位点的细胞类型组成,可选择不同模式预测位点内1-2种或多种细胞类型。此外,RCTD还开发了一套均一化的方法,通过结合基因特异性随机效应项来解决不同建库测序方法带来的平台效应。
-
【文献案例一】

-
期刊:Journal of Translational Medicine
-
影响因子:6.1
-
单细胞技术:数据库scRNA数据;10x空间转录组
增生性结节形成是良性前列腺增生(BPH)的一个特征性病理特征,是前列腺体积增大和随之而来的下尿路症状(LUTS)的主要原因。其具体机制在很大程度上尚不清楚,尽管有报道称几个细胞过程参与了BPH的发生和发展,并强调了上皮细胞在增殖性结节形成中的关键作用。然而,技术限制阻碍了BPH患者的体内研究。本文采用稳健细胞类型分解(RCTD)方法整合空间转录组学和单细胞RNA测序图谱,从而阐明结节形成过程中上皮细胞的改变。
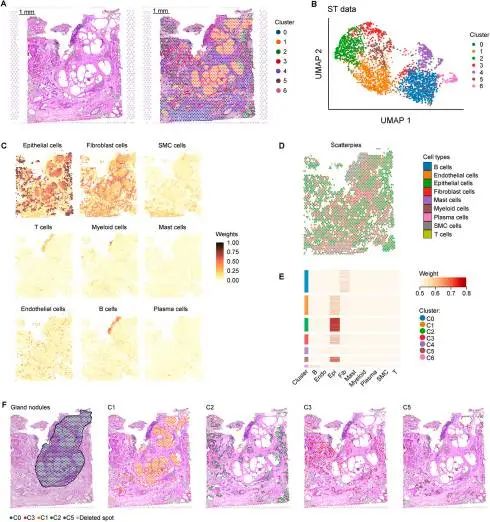
04 Cell2location
空间转录组的spot中往往含有多种细胞类型;另外,空间组学数据成分多样化,例如不同组织位置中细胞数量的差异,不同细胞类型中总mRNA的差异,以及切片厚度差异导致获得的细胞成分比例不同等。Cell2location【5】在开发时考虑到这些因素,基于贝叶斯模型,能够达到比其他方法更高的细粒度(fine-grained classification),从而全面地在不同组织中定位各种细分的细胞类型。
-
【文献案例一】

-
期刊:Nature Genetics
-
影响因子:31.7
-
单细胞技术:10x scRNA数据;10x空间转录组
子宫内膜功能障碍是许多常见疾病的基础,包括异常子宫出血、不孕症、流产、先兆子痫、子宫内膜异位症和子宫内膜癌,这些疾病共同影响着世界各地的许多女性。解剖在人类月经周期中调节细胞分化的机制对于理解正常子宫内膜如何被调节是至关重要的。本文生成了人类子宫和三维子宫内膜类器官的单细胞和空间图谱,阐明了子宫内膜癌和子宫内膜异位症病变的细胞类型。文章利用cell2location定义了三个以上皮细胞为中心的子宫内膜微环境,并揭示了上皮-基质之间的相互作用。
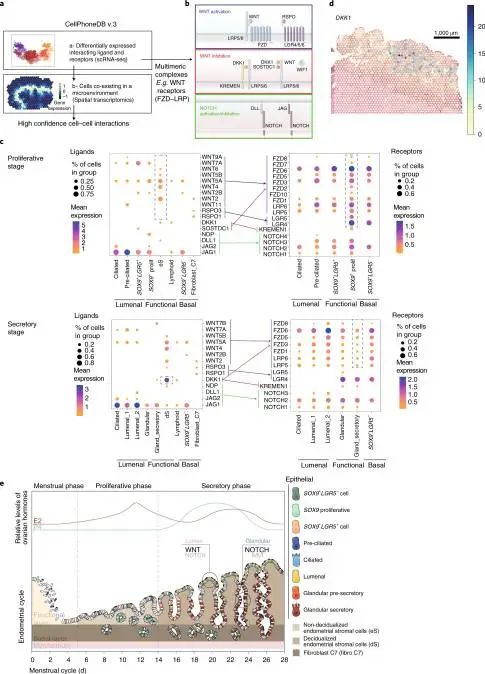
二、利用scRNA映射(mapping)对空间转录组进行注释
scRNA mapping主要有2种方式,将指定的基于scRNA的细胞亚型定位到HPRI(high-plex RNA imaging)图谱上的每个细胞,以及将每个scRNA-seq细胞定位到组织的特定区域。
01 Seurat mapping
Seurat是10x官方开放出来用于分析单细胞数据(scRNA以及空间转录组都能分析)的软件,目前已更新到了5.0版本。Seurat提供了两种工作流来识别与组织内空间位置相关的分子特征。第一种是基于组织内预注释的解剖区域进行差异表达,这些区域可以通过无监督聚类或先验知识确定。第二种是在没有预注释的情况下,通过FindSpatiallyVariables寻找空间模式的特征。
-
【文献案例一】

-
期刊:Proceedings of the National Academy of Sciences of the United States of America
-
影响因子:9.4
-
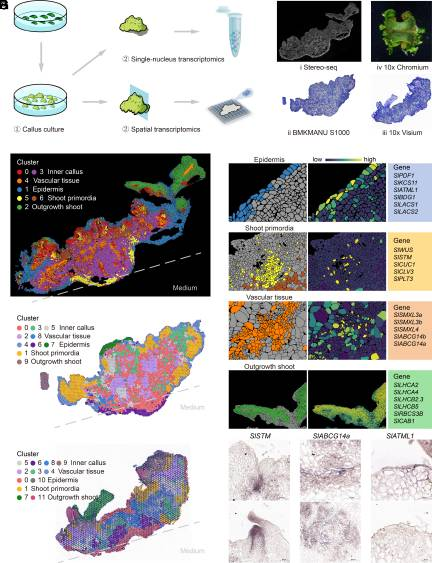
单细胞技术:10x scRNA数据;10x空间转录组;Stereo-seq;BMKMANU S1000
愈伤组织是一种在作物工程中参与植物再生和基因转化的重编程细胞团。多能愈伤组织通过芽再生发育成可育芽。作物愈伤组织中茎再生过程的分子基础在很大程度上仍然难以捉摸。本研究对番茄愈伤组织再生过程中的空间转录组(多种空转手段)进行了初步探索。文章使用seurat对空转数据进行了聚类,并原位呈现出了7个cluster的精确空间分布。注释结果与解剖观察结果高度吻合,并且三种空间转录组测序结果具有一致性。
02 celltrek
CellTrek发表于2022年的Nature Biotechnology【6】,其能够将空间转录组和scRNA的原始矩阵合并及共降维,随后利用其中的空转数据构建多元随机森林模型(RF),将scRNA里的单个细胞映射到空间坐标上。空转的spot往往达不到单细胞的水平,并且去卷积方法是将空转数据作为bulk RNA计算细胞比例,更适合定性。而cellTrek这种映射不仅可以实现单细胞水平,同时还能够进行准确定量以及进行后续更精细化的分析。
-
【文献案例一】

-
期刊:Journal of dental research
-
影响因子:5.7
-
单细胞技术:10x scRNA-seq;stereo-seq
腭裂是人类最普遍的出生缺陷之一,不幸的是,腭裂的早期筛查仍然是一个挑战,即使是先进的技术,如三维产前超声,也无法提供可靠的唇裂和/或腭裂诊断。目前缺乏整个上颚发育过程中空间单细胞分辨率的完整图谱,阻碍了对复杂基因调控过程的全面理解。本文利用CellTrek整合了scRNA-seq和stereo-seq在腭增殖期(E12.5)、腭融合期(E14.5)和腭发育完成期(E16.5)的数据集,建立了小鼠腭发育过程中的转录组调控景观。
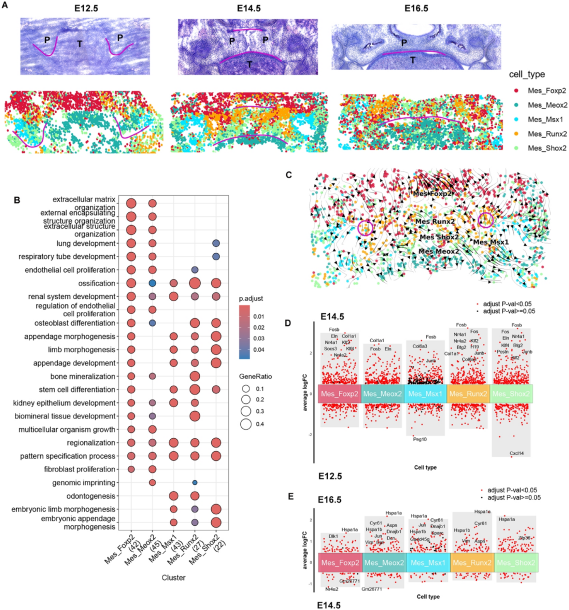
-
【文献案例二】

-
期刊:Molecular Cancer
-
影响因子:27.7
-
单细胞技术:数据库公开scRNA与ST数据;
癌症相关成纤维细胞(CAFs)是一种异质性细胞群,在重塑肿瘤微环境(TME)中起着至关重要的作用。目前,包括免疫治疗和化疗在内的大多数治疗方法在很大程度上忽视了CAFs的关键作用。对CAFs和TME成分之间相互作用的理解不足以支持可靠治疗策略的发展。在这项研究中,作者描绘了六种常见癌症类型中CAF的格局,并描述了这些亚型的独特功能特征。通过整合scRNA-seq数据和ST数据生成了跨越6个肿瘤的空间单细胞转录组图谱,用于描述CAFs的空间分布特征,并表征CAFs与TME之间的复杂相互作用。本文利用celltrek将scRNA的数据映射到了ST空间图谱中,最终获得了包含744,289个细胞的泛癌空间单细胞转录图谱。
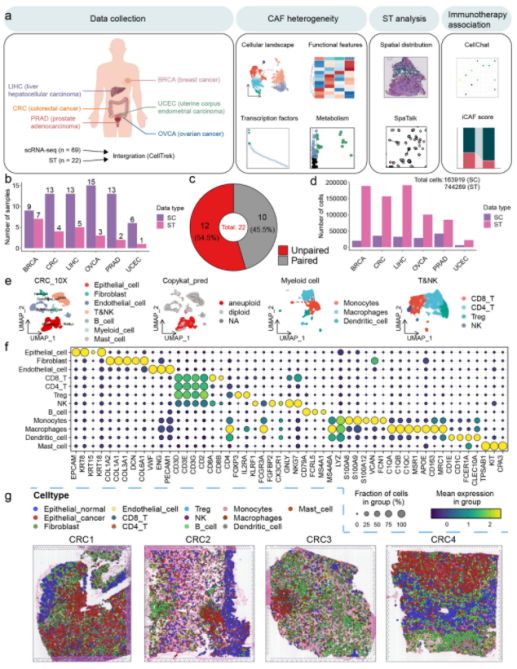
03 SPANN
2024年1月,北京大学邓明华教授团队在Briefings in Bioinformatics上发表了一篇文章SPANN: annotating single-cell resolution spatial transcriptome data with scRNA-seq data【7】,并带来了利用scRNA-seq数据来注释单细胞分辨率的空间转录组数据的新方法SPANN。此方法相对于其他算法,不仅保留了已知细胞类型注释的高精度,还支持自动检测未知细胞类型。
文章展示了使用不同算法对胚胎seqFISH与scRNA数据集进行映射的结果。在对齐之前,两个数据集的UMAP可视化之间几乎没有重叠。这表明了在scRNA-seq和空间转录组数据之间进行比对的必要性。而在映射后,SeqFISH簇可分离性较好,细胞类型对应关系较为明确。并且相对于其他算法而言(如Seurat、Liger、GLUE和Uniport),SPANN拥有更紧凑的聚类和更少的异常值。
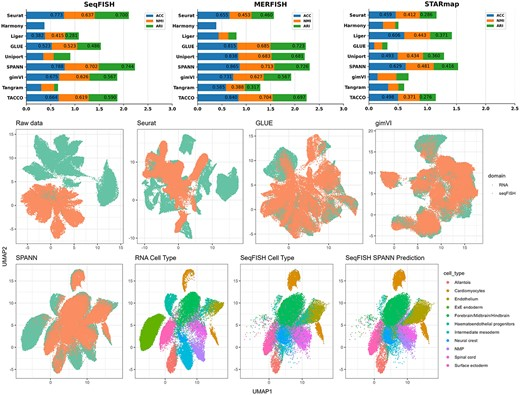
除了以上展示的基于“Deconvolution”以及“Mapping”的几种算法,实际还有很多其他的软件也能达到scRNA+ST数据联合注释的效果,如gimVI、Harmony等等。感兴趣的老师也可以仔细去研读一下前面所提到的斯坦福大学发的那篇文章,这里就不一一介绍啦。
「 题外话一 」
目前空间转录组的产品很多,例如10x Genomics的 Visium相关产品、华大的stereo-seq、百迈客的BMKMANU S1000、美国Broad研究所开发的slide-seq等等。其中stereo-seq的文章也在逐年增加,高分辨率以及自由定制芯片的特点也受到很多老师青睐。不同的注释方法在stereo-seq数据中效果如何,是否适合在stereo-seq数据中使用,可以去详细看一下这篇文章【Benchmarking mapping algorithms for cell-type annotating in mouse brain by integrating single-nucleus RNA-seq and Stereo-seq data】【8】。
「 题外话二 」
没有scRNA-seq数据能不能做空间转录组数据?答案当然是肯定的。例如直接利用seurat进行空间转录组的聚类;基于数据自身cluster定义的marker gene ,结合生物学背景来注释spot细胞类型;或者使用BayesPrism、Bulk2Space这类利用bulk RNA数据解卷积的算法来对空间转录组数据进行注释。但广州国家实验室田鲁亦团队联合西湖大学刘晓东团队合作发表的文章“Systematic comparison of sequencing-based spatial transcriptomic methods”【9】表明scRNA的细胞注释(单细胞维度)对于提升空间转录组注释的精确度(特别是解析具有空间模式的稀有细胞状态方面)非常重要。因此,对于有科研条件、并且看重高质量分析数据的科研团队而言,联合scRNA与空间转录组是十分有必要的。
-
参考文献
1、Longo, S.K., Guo, M.G., Ji, A.L. et al. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat Rev Genet 22, 627–644 (2021). https://doi.org/10.1038/s41576-021-00370-8
2、Elosua-Bayes M, Nieto P, Mereu E, Gut I, Heyn H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 2021 May 21;49(9):e50. doi: 10.1093/nar/gkab043. PMID: 33544846; PMCID: PMC8136778.
3、Ma Y, Zhou X. Spatially informed cell-type deconvolution for spatial transcriptomics. Nat Biotechnol. 2022 Sep;40(9):1349-1359. doi: 10.1038/s41587-022-01273-7. Epub 2022 May 2. PMID: 35501392; PMCID: PMC9464662.
4、Cable DM, Murray E, Zou LS, Goeva A, Macosko EZ, Chen F, Irizarry RA. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat Biotechnol. 2022 Apr;40(4):517-526. doi: 10.1038/s41587-021-00830-w. Epub 2021 Feb 18. PMID: 33603203; PMCID: PMC8606190.
5、Kleshchevnikov V, Shmatko A, Dann E, Aivazidis A, King HW, Li T, Elmentaite R, Lomakin A, Kedlian V, Gayoso A, Jain MS, Park JS, Ramona L, Tuck E, Arutyunyan A, Vento-Tormo R, Gerstung M, James L, Stegle O, Bayraktar OA. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat Biotechnol. 2022 May;40(5):661-671. doi: 10.1038/s41587-021-01139-4. Epub 2022 Jan 13. PMID: 35027729.
6、Wei R, He S, Bai S, Sei E, Hu M, Thompson A, Chen K, Krishnamurthy S, Navin NE. Spatial charting of single-cell transcriptomes in tissues. Nat Biotechnol. 2022 Aug;40(8):1190-1199. doi: 10.1038/s41587-022-01233-1. Epub 2022 Mar 21. PMID: 35314812; PMCID: PMC9673606.
7、Yuan, M., Wan, H., Wang, Z., Guo, Q., & Deng, M. (2024). SPANN: annotating single-cell resolution spatial transcriptome data with scRNA-seq data. Briefings in Bioinformatics, 25(2), bbad533.
8、Quyuan Tao, Yiheng Xu, Youzhe He, Ting Luo, Xiaoming Li, Lei Han, Benchmarking mapping algorithms for cell-type annotating in mouse brain by integrating single-nucleus RNA-seq and Stereo-seq data, Briefings in Bioinform https://doi.org/10.1093/bib/bbae250
9、You Y, Fu Y, Li L, Zhang Z, Jia S, Lu S, Ren W, Liu Y, Xu Y, Liu X, Jiang F, Peng G, Sampath Kumar A, Ritchie ME, Liu X, Tian L. Systematic comparison of sequencing-based spatial transcriptomic methods. Nat Methods. 2024 Jul 4. doi: 10.1038/s41592-024-02325-3. Epub ahead of print. PMID: 38965443.

















 580
580

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









