











研究背景
质子交换膜燃料电池(PEMFCs)被认为是一种很有前途的清洁和可持续的能源转换设备。然而,其应用受到ORR缓慢动力学的限制。Pt/C是目前最常见的商用ORR催化剂,但其成本高、耐久性差等缺点阻碍了其大规模使用。因此,迫切需要寻找一种活性高、低成本、长期耐久性好的ORR催化剂来取代Pt/C。
为了提高氧还原反应(ORR)的催化活性,M−N−C材料中广泛采用P掺杂。然而,P掺杂的ORR机制尚不清楚。天津大学刘恩佐等人基于第一性原理的计算,研究了P掺杂FeN4的稳定性和ORR活性,并通过微动力学模型确定了表面覆盖与电势的关系,该方法也可应用于其他杂原子掺杂单原子电催化剂的研究。
计算方法
本文采用vasp软件包对其催化性能及电子性质进行理论研究,计算基于广义梯度近似(GGA)下的PBE泛函计算电子交换相关性,离子-电子相互作用采用PAW方法进行表征。所有计算都考虑自旋极化。
利用Grimme的DFT-D3方案来描述长程氢键相互作用,Z方向真空度设置为20 Å,截断能设置为520 eV,将最大力的收敛公差设为0.01 eV·Å-1。根据Fe和P含量的实验表征,采用Fe和P的1:1化学计量比构建了掺杂P的FeN4模型。利用CI-NEB对过渡态(TS)进行识别。
结果与讨论
为了研究P掺杂对FeN4 对ORR过程的影响,作者首先研究了7个可能的掺杂位点(包括1个N位点和6个C位点,图1a),P掺杂的形成能总结在图1b中。从图1b中可以看出,P3是能量最有利的掺杂位点,其次是N3P。在确定了最稳定的掺杂构型(FeN4P3)后,我们开始研究p掺杂效应,重点研究了P掺杂引发ORR中间体吸附行为的变化。这些变化主要体现在两个方面:(1)外来磷对中间产物在Fe位点的吸附行为的影响;(2)中间产物在P位点的吸附行为。
在Fe位点上的吸附行为将在后面讨论。我们讨论了P位点的中间吸附,比较了P和C位点的差异。图1c显示了P3和C3位点的ORR自由能。可以看出,OH中间体在P3位点的解吸受到了强烈的阻碍(实际上,O的质子化也很困难)。与P3位点相反,C3位点的OH解吸非常容易,而OOH的形成则受到阻碍。从图1d可以看出,其他P和C位点的GOH*值与P3和C3位点相似,均远未达到最佳吸附强度。因此P和C不是ORR活性位点。

图1 P掺杂催化剂结构及OER性能关系
P掺杂FeN4结构的催化剂在实现OER过程中需要考虑中间体在其表面的覆盖程度。图2(a)~(c)分别为FeN4、FeN3PO和FeN3P模型吸附特征中间体在外加电压下的覆盖程度。图2e为考虑P点覆盖度和不考虑P点覆盖度时Fe点ORR的理论起始势,FeN4PX (FeN3P)和FeN4PXO (FeN3PO)之间存在不可忽略的差异,这说明即使研究Fe点的ORR过程,P点的覆盖也不能忽略。
随着电位的增加,反应速率减慢,物种覆盖范围由*Fe依次变化为OH*Fe和O*Fe(图2f)。换句话说,随着电位的增加,OH*的去除和O*的氢化作用依次被中断,因此OH*Fe和O*Fe是Fe位置的主要覆盖物种。
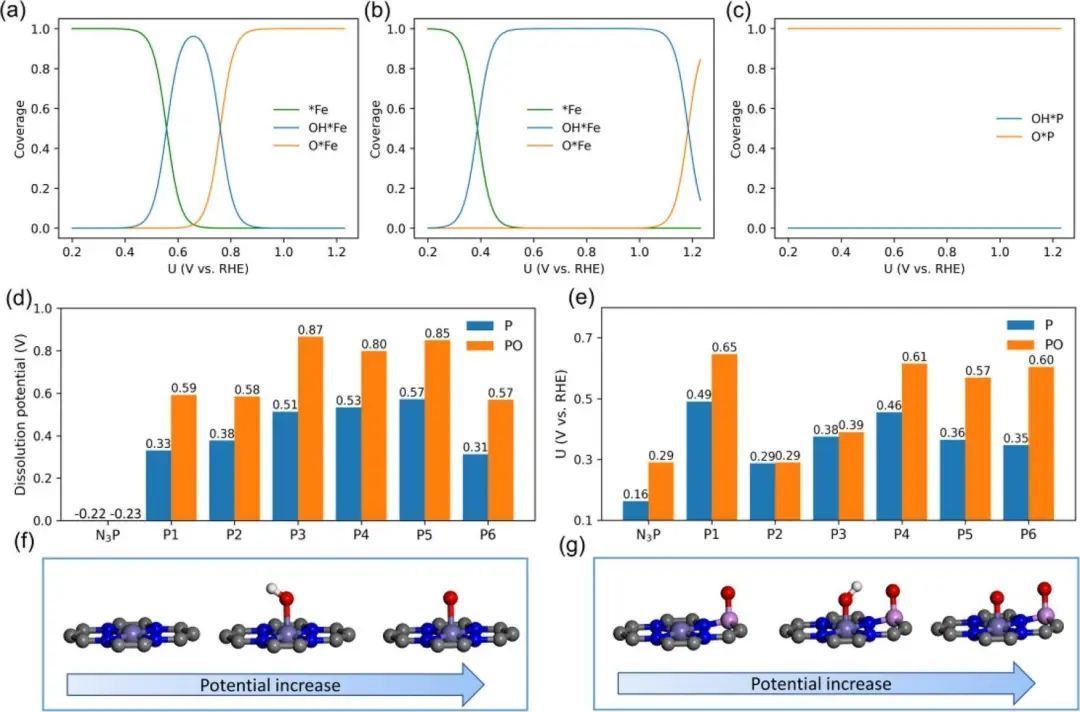 图2 (a) FeN4模型中Fe位点的物种覆盖度。(b) FeN4P3O模型中Fe位点的物种覆盖度。(c) FeN4P3模型中P点的物种覆盖度。(d) P掺杂FeN4的溶解势。P的标记(蓝色)代表裸P, PO(橙色)代表O原子覆盖的P位点。(e) P掺杂FeN4的理论起始电位(Uonset)。(f) FeN4随势增加的物种覆盖模型。(g) FeN4P3O随电位增加的物种覆盖模型。
图2 (a) FeN4模型中Fe位点的物种覆盖度。(b) FeN4P3O模型中Fe位点的物种覆盖度。(c) FeN4P3模型中P点的物种覆盖度。(d) P掺杂FeN4的溶解势。P的标记(蓝色)代表裸P, PO(橙色)代表O原子覆盖的P位点。(e) P掺杂FeN4的理论起始电位(Uonset)。(f) FeN4随势增加的物种覆盖模型。(g) FeN4P3O随电位增加的物种覆盖模型。
采用HCA模型来讨论了FeN4和FeN4PO的ORR活性。为了研究P掺杂的ORR机制,我们基于FeN4和FeN4P模型绘制了ORR路径,分别如图3a和3b所示。FeN4有3条与Fe位点对应的反应路径,如图3a所示,即FeN4(Path 1)、FeOHN4(Path 2)和FeON4(Path 3)。路径1在该电位(0 ~ 0.6 V)下进行,ORR起始电位为0.56 V。随着电位的增大(0.4 ~ 0.8 V),电化学步骤的反应速率减慢,游离*Fe逐渐转变为OH*Fe(图2a),形成FeOHN4构型。进行路径2,起始电位为0.84 V。
随着电势进一步增大(0.7 ~ 1.2 V),最终形成FeON4;执行路径3,起始电位为0.61 V。从FeN4的整个路径图(图3a)可以看出,如果FeOHN4(路径2,图3a)的电位范围能够扩大,将非常有利于高电位下的ORR过程。作者考虑了多氧态P的形成,如图3c所示。可以看出,PO到PO4的速率决定步骤(RDS)是第一步,即PO到PO2的转换。负自由能变化意味着PO可以自发形成PO4,但这一过程将受到Fe位的竞争。FeOPO构型在能量上比PO2更稳定,FeOPO2构型比PO3更稳定。
同时,POX的质子化过程非常困难。因此,形成PO4的主要障碍是Fe位点对O原子的竞争。PO取代PO4成为主要构型,而且PO4的形成伴随着Fe位的激烈竞争。热力学上,FeOHN4PO和FeN4PO4都是比FeN4OH更好的ORR活性位点。
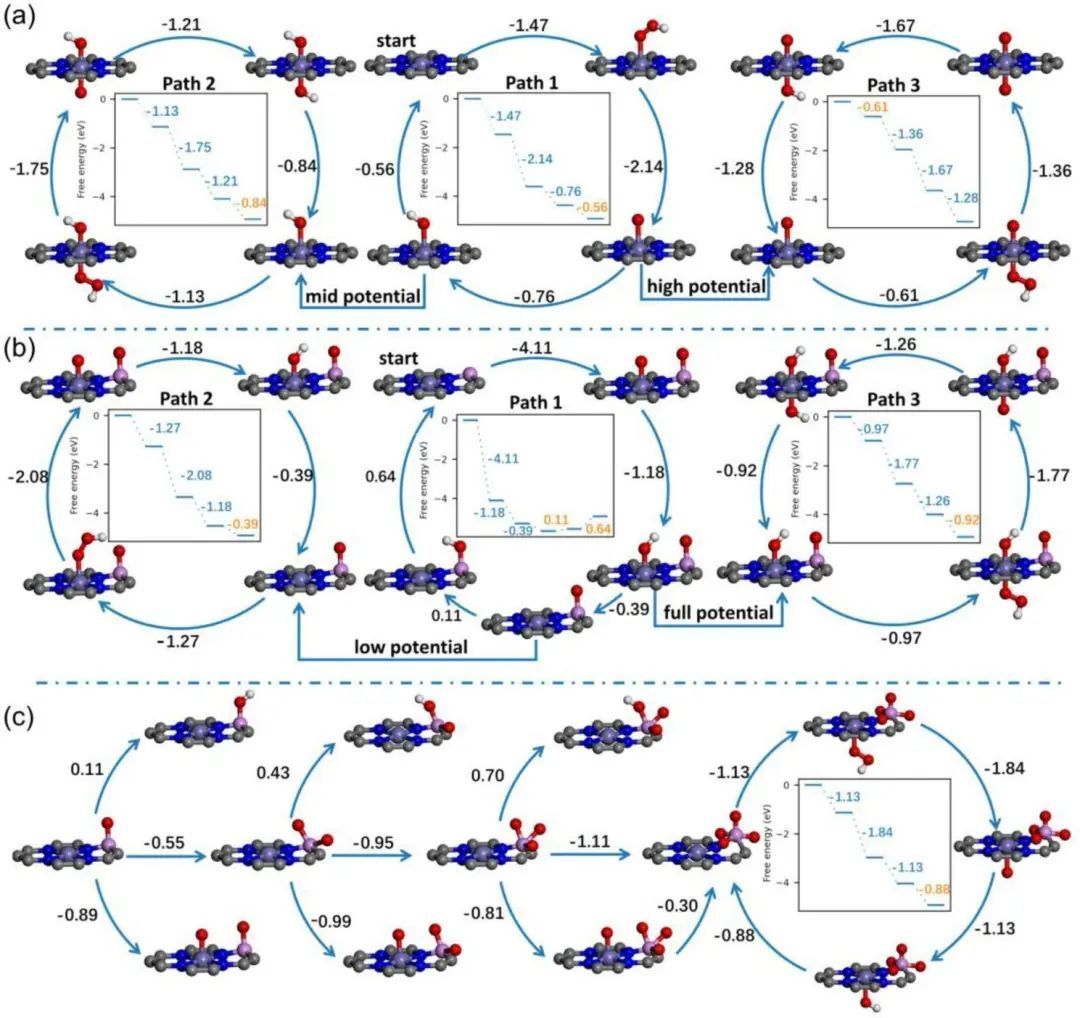
图3 (a) FeN4和(b) FeN4P的反应途径图和ORR自由能。(c)FeN4PO4 ORR自由能过程
轴向OH配体范围的拓宽根源于稳定OH*Fe−O*P之间的氢键,如图4a所示,OH*Fe−O*P的键长为1.955 Å。图4b显示了H-1s和O- 2p轨道的态密度(DOS)和COHP。COHP用于计算电子状态重叠对成键或反键轨道的贡献。图4b显示了H−O的s−p轨道杂化和负iCOHP(−0.55 eV),证明了长程氢键相互作用。氢键促进机制稳定了Fe位上的OH中间体,从而拓宽了FeOHN4PO活性结构的范围。
从图4c可以看出,这种稳定效应可以打破ΔG3−ΔG4的缩放关系,这意味着Fe位点(在FeN4P3O中)由于ΔG3的负电荷较多,更容易保持OH的覆盖,而远离O的覆盖。此外,根据Fe位的Bader电荷分析, FeN4 (1.101 e-)转变FeOHN4 (1.229 e-)和FeN4PO (1.136 e-)转变FeOHN4PO (1.309 e-),表明OH轴向配体由于O的电负性增强而增强,增强了Fe的价态,削弱了OH*的吸附,促进了ORR过程。因此,催化活性结构需要对ORR中间体有适度的吸附。
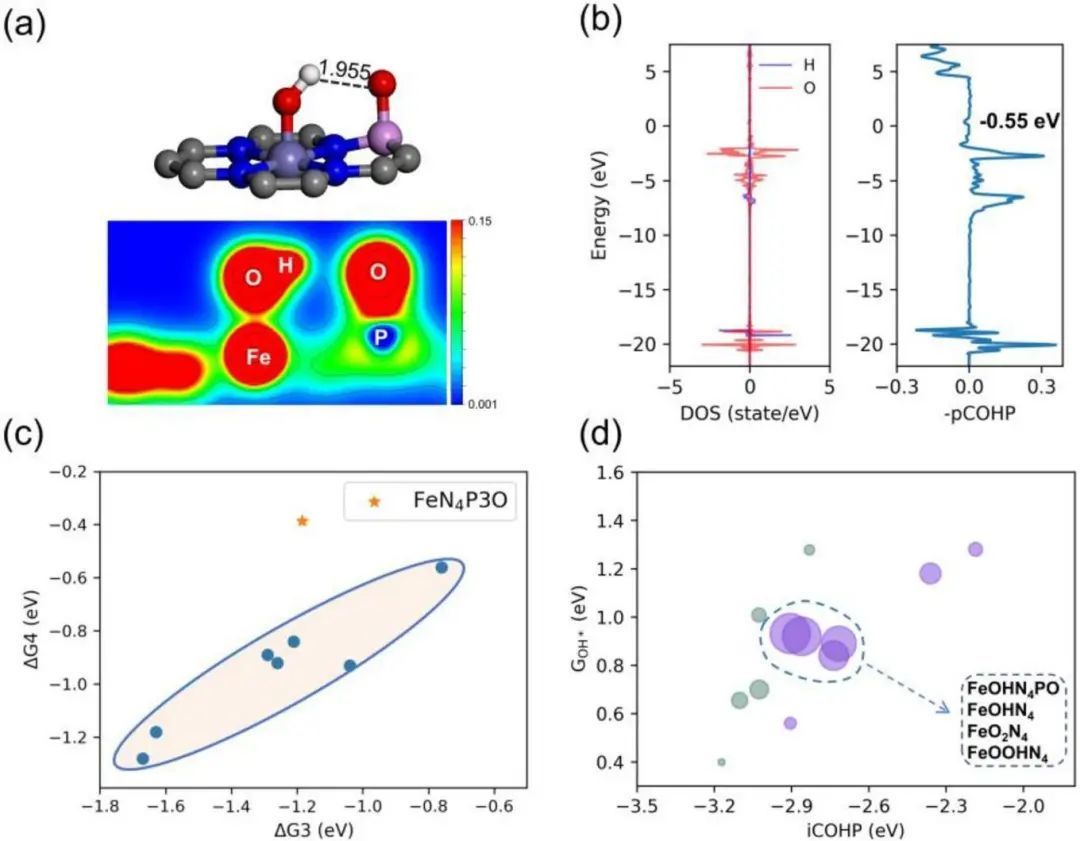
图4 催化剂电子结构及自由能关系
为了更全面地了解氢键促进机制,特别是扩大OH覆盖范围对ORR动力学的促进作用,利用微动力学模型模拟了各个活性位点的极化曲线。首先是FeN4的三个路径的模拟(图3a),分别对应三个活性结构(FeN4, FeOHN4, FeON4),如图5a所示。括号内标出FeN4的潜在中间体覆盖范围。FeOHN4结构具有最高的半波电位(0.95 V),其次是FeN4 (0.68 V),最后是FeN4O (0.57 V), FeOHN4具有最高的半波电位,证明FeOHN4具有优异的ORR催化活性。以FeN4为活性基准,FeON4会阻碍ORR动力学,而FeOHN4会加速ORR动力学。FeN4PO的模拟极化曲线如图5b所示。
FeOHN4PO具有最高的半波电位(0.96 V),其次是FeON4PO (0.71 V),最后是FeN4PO (0.51 V),与FeOHN4相似,FeOHN4PO也具有最高的ORR半波电位。对比Fig . 5a和Fig . 5b,可以发现FeOHN4PO的半波电位略高于FeN4OH,可见P掺杂的关键促进作用在于扩大OH覆盖,而不是增强位点活性。
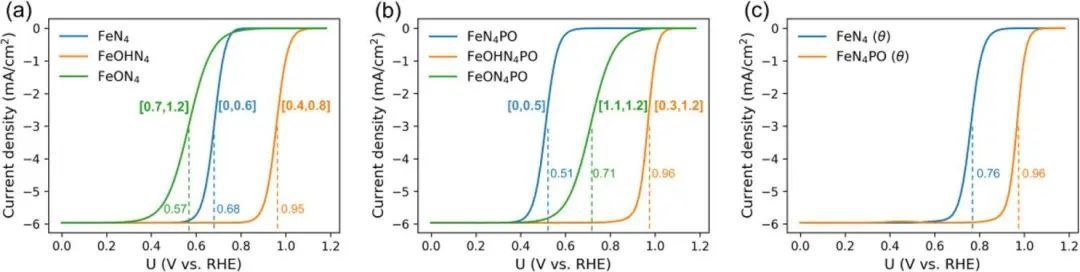
图5 模拟了FeN4、FeOHN4和FeON4的极化曲线和动力学电流密度
结论与展望
本工作详细研究了P掺杂对Fe - N - C材料ORR性能的影响,包括优化的掺杂位置、电化学稳定性以及增强的ORR热力学和动力学,揭示了氢键促进的ORR机制。本工作为催化机制提供了一个有深刻见解的视角。同时,方法可以推广到其他掺杂剂的研究,如S掺杂,并指导单原子电催化剂的设计。
文献信息
Li, B., Shi, C., Zhao, N., & Liu, E. (2023). Hydrogen-Bond-Promoted ORR Mechanism in P-Doped Fe–N–C Materials. The Journal of Physical Chemistry C.ht-tps://doi.org/10.1021/acs.jpcc.2c07160















 2926
2926











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









