










 研究者通过高通量筛选和第一性原理计算,选出了Cs2KMI6(M=Ga,In)作为有潜力的新型无铅钙钛矿,探讨了其热稳定性和光电性能,发现K-I键为共价,M-卤素键和Cs-I键为离子键,可用于高效光电器件设计。
研究者通过高通量筛选和第一性原理计算,选出了Cs2KMI6(M=Ga,In)作为有潜力的新型无铅钙钛矿,探讨了其热稳定性和光电性能,发现K-I键为共价,M-卤素键和Cs-I键为离子键,可用于高效光电器件设计。

研究背景
开发具有合适带隙和优越热稳定性的新型无铅钙钛矿材料,对于提高其在下一代光伏技术中的应用至关重要。高通量筛选与第一原理方法相结合,可以准确有效地筛选出有前景的钙钛矿。在此,福建物质结构研究所卢灿忠和孟令一等人从1026个化合物中选择了两种无铅全无机卤化物双钙钛矿材料Cs2KMI6(M = Ga,In),其标准包括适当的结构因子、正分解能和合适的直接带隙。本文研究了两种钙钛矿Cs2KMI6(M = Ga,In)的热稳定性和力学稳定性、几何结构和电子结构、光电性能和缺陷形成能。通过对结构因子、弹性常数和稳定的化学势区域的分析,探究它们的结构成形性和稳定性,并对其光电性能进行分析。
计算方法
在这项工作中,通过VASP量子计算软件包进行第一性原理的理论计算。1026个钙钛矿的初始晶体结构,空间群为Fmm,选择两种钙钛矿Cs2KMI6(M = Ga,In),采用广义梯度近似(GGA)中的PBE泛函数来描述交换相关离子效应。k点网格设置为3×3×3为几何优化,6×6×6为自洽计算。能量和最大力移的收敛率分别设置为2×10−6 Ha和0.01eV/Å,截断能为520 eV。同时考虑到PBE泛函通常被低估的带隙,采用PBE+SOC(自旋轨道耦合)、HSE06和HSE06 + SOC泛函来计算能带结构。
结果与讨论
如图1所示,作者通过结合4种不同的A位阳离子(Cs+、K+、Na+、Rb+)、Rb+ )、10M(I)位点的一价阳离子(Ag+,Au+,Cu+,In+,K+,Li+,Na+,Rb+,Tl+,和Cs+),,9M(III)位点三价阳离子(Al3+,As3+、Au3+、Bi3+、Ga3+,In3+、Sb3+、Sc3+和Y3+),4个X位阴离子(X = F、Cl、Br、I),共生成了1026个A2M(I)M(III)X6化合物。通过高通量筛选和DFT计算,对1026种化合物中具有良好结构稳定性和合适的直接带隙的无机无铅双钙钛矿进行了评价。首先,结构因子(如耐受性因子和八面体因子)是获取稳定钙钛矿结构形成性的适当筛选标准。如图1(a)所示,A2M(I)M(III)X6双钙钛矿由交替的M (I)X6和M(III)X6八面体组成,其结构稳定性和成形性与组成离子的半径有关。通过进一步排除含有有毒元素(如Tl和As)的化合物,作者筛选了8个候选化合物,包括Cs2KGaI6、Cs2KInI6、Cs2RbGaI6、Rb2AuAlCl6、K2AuAlCl6、K2AgGaCl6、K2AgInCl6和Rb2AgInCl6。最后,作者用第一性原理方法系统地研究Cs基钙钛矿Cs2KGaI6和Cs2KInI6的热稳定性、电子结构、光电性质和缺陷性质。Cs2KGaI6和Cs2KInI6的计算结构因子和预测分解能见表1。
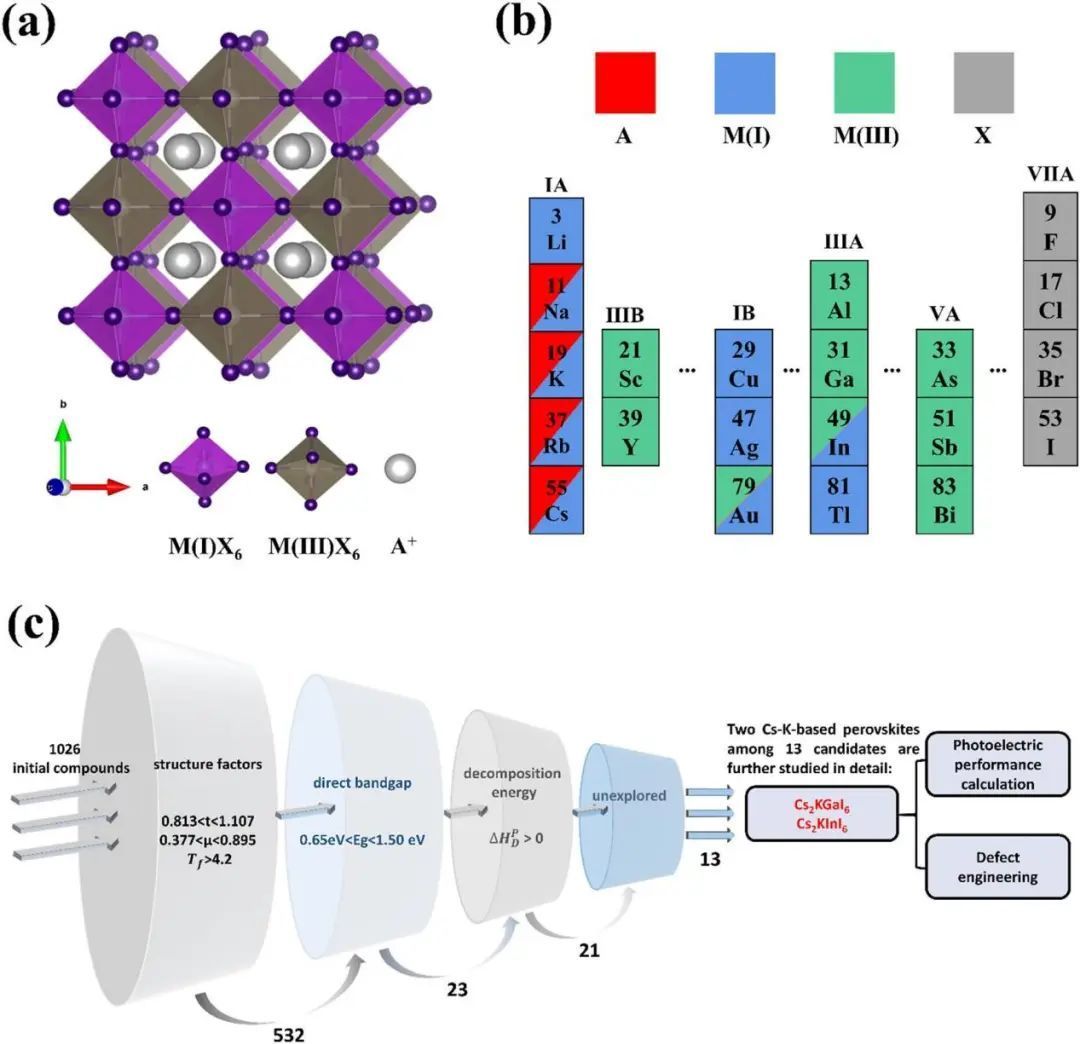
图1 (a) A2M(I)M(III)X6双钙钛矿的晶体结构;(b) A2M(I)M(III)X6化合物中考虑的元素,A位点、M(I)位点、M(III)位点和X位点分别标记为红色、蓝色、绿色和深灰色;(c)无铅无机双钙钛矿化合物的筛选流程图
为了进一步评价钙钛矿的热稳定性,根据第一原理方法计算了稳定性相图。计算得到的Cs2KGaI6和Cs2KInI6的生成能分别为-13.54 eV和-13.82 eV,表明体系可以稳定存在。此外,作者分析了所有实验合成的钙钛矿Cs2KMI6(M = Ga,In)对应的二元和三元化合物,并计算了这些相的稳定化学势区域,绘制了钙钛矿Cs2KMI6(M = Ga,In)的热力学相图,如图2所示。这些现有的热力学相图可以用来分析Cs2KMI6(M = Ga,In)的热稳定性。狭窄的二维多面体区域(如图2所示)表明,需要仔细控制热力学合成条件,以获得高质量的Cs2KMI6(M = Ga,In),避免杂质相的产生。根据计算得到的热力学稳定相图,我们选择了两个具有代表性的化学条件,分别对应于点A/C点(Ga/In-rich,I-poor)和B/D点(Ga/In-poor,I-rich)的化学条件,如图2所示。一般来说,形成能较高的缺陷意味着其成形性较低。因此,在接下来的讨论中,我们将重点关注具有低缺陷形成能量的缺陷,这对化合物的晶体生长和光电性能有很大的影响。发现两种化合物都存在某些类型的缺陷。例如,在富M或贫M化学条件下,供体型缺陷包括间隙缺陷Mi和Csi,而受体型缺陷包括涉及M元素的取代缺陷,如CsM、IM和KM,因此缺陷的形成可以通过调整化学条件来控制。
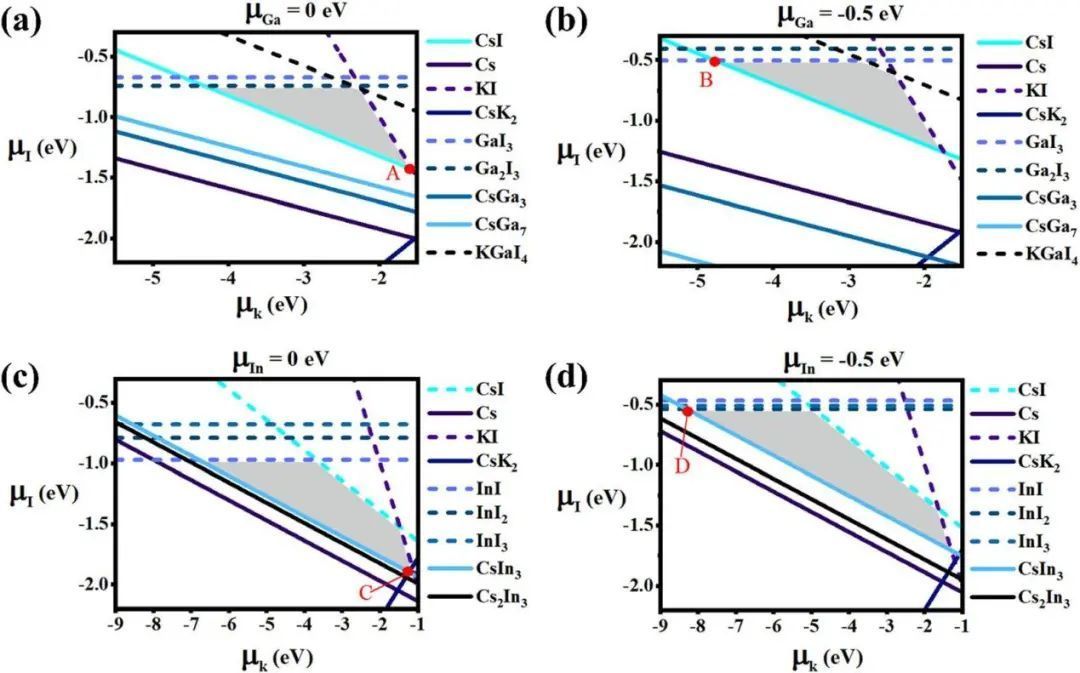
图2 催化剂热力学相图
众所周知,电子结构与半导体材料的光伏性能密切相关。利用PBE、PBE + SOC、HSE06和HSE06 + SOC计算了Cs2KMI6(M = Ga,In)的带隙,如表2和图3所示,并展示了两种化合物的直接带结构,PBE带隙与数据库一致。如图3所示,价带边和导带边(对应于G点)是色散的,这可能导致相对较小的有效质量和较大的载流子迁移率。当元素从Ga变为In时,导带向上偏移,从而导致带隙增大。根据Shockley–Queisser极限,带隙为0.8-2.2eV的钙钛矿材料广泛应用于光伏转换领域。作为比较,先前报道的钙钛矿Cs2AgBiBr6表现出更大的间接带隙(例如与HSE06功能的2.21eV),其计算的电子有效质量(L-G:0.58m0和L-W:0.34m0)大于Cs2KMI6(M = Ga,In)。
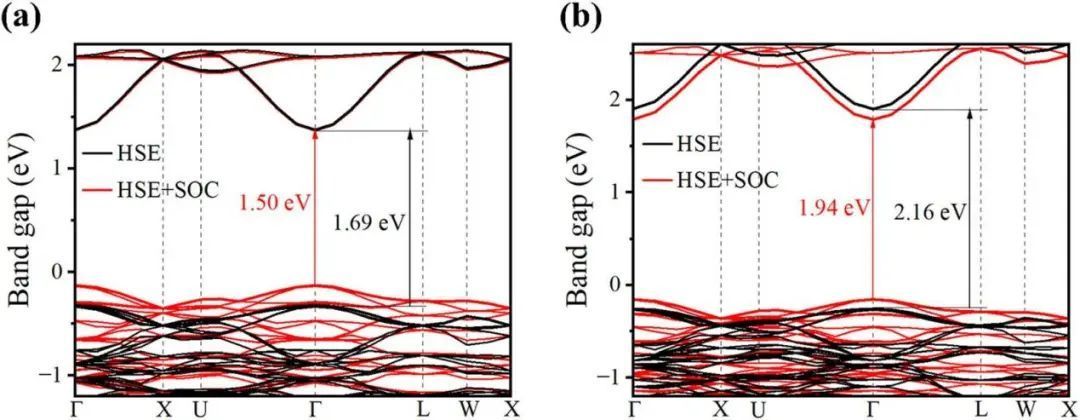
图3 能带结构图
ELF可以用来可视化原子壳结构,键对的分布以及分子中的电子孤对,从而监测键形成和断裂过程中电子分布的变化。较小的ELF值意味着电子在两个原子之间具有超低的密度,具有典型的离子键特征。相反,当ELF接近1时,电子以高密度定域在两个原子之间,这表明存在一个理想的共价键。ELF计算可以清楚地显示金属阳离子(如Cs+、K+和M3+)与I之间的键合性质。如图4所示,M和I离子中心之间的电子共享行为表明它们之间的共价键越多K+、Cs+和其他离子之间较低的ELF值表明K、Cs和其他离子之间的离子相互作用,类似于Cs、K和[MI6]单元之间的离子相互作用。这些原子表明Cs和K原子通常作为电荷供体。M和I离子之间的耦合明显强于K和I离子之间的耦合,导致K-I键长增大。此外,还进行了Bader电荷分析,如图4所示。正的Bader电荷表示原子在电荷转移中提供电荷,而负电荷表示原子接受电荷。这进一步证明了Cs和K原子作为电荷供体。先前的研究表明,钙钛矿中增强的电荷转移意味着具有良好的稳定性。因此,Cs2KInI6的电荷转移中的Bader电荷略大于其他化合物,说明Cs2KInI6的稳定性略好。
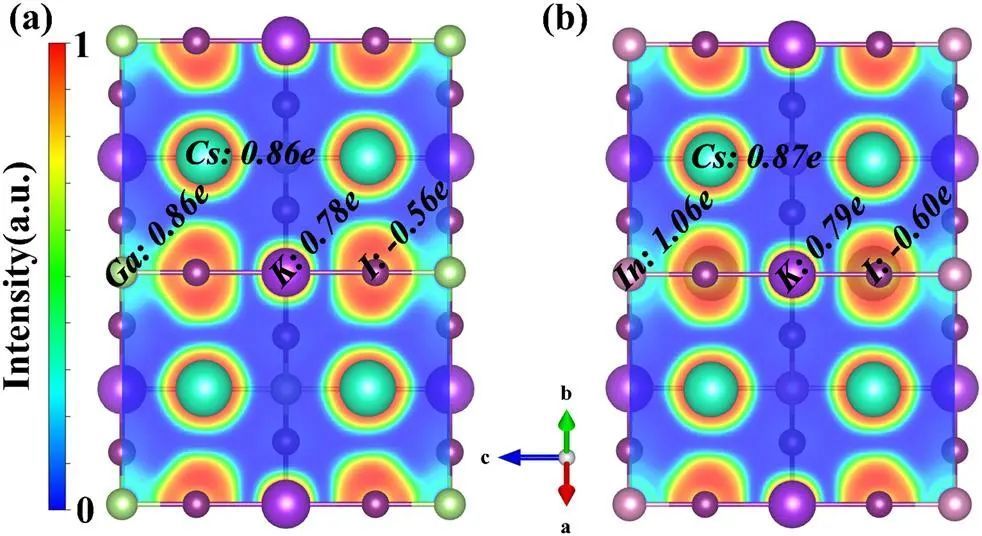
图4 电子局域函数及Bader电荷
有效的光吸收在光电应用中具有重要意义,它是由光与材料之间的相互作用决定的。通过HSE06计算了Cs2KMI6(M = Ga,In)的光吸收系数(见图5(a)),并分析了这些钙钛矿在可见光区域的吸收性能。Cs2KMI6(M = Ga,In)在可见光区域的吸收系数高达105 cm -1。SLME是一种评估光伏器件性能的常用工具。与Shockley–Queisser极限相比,SLME不仅考虑了能带结构,还考虑了活性膜的厚度和吸收系数。如图5(b)所示,Cs2KGaI6的SLME分别达到28.39%(膜厚为530 nm),Cs2KInI6的SLME分别为19.05%(480 nm)
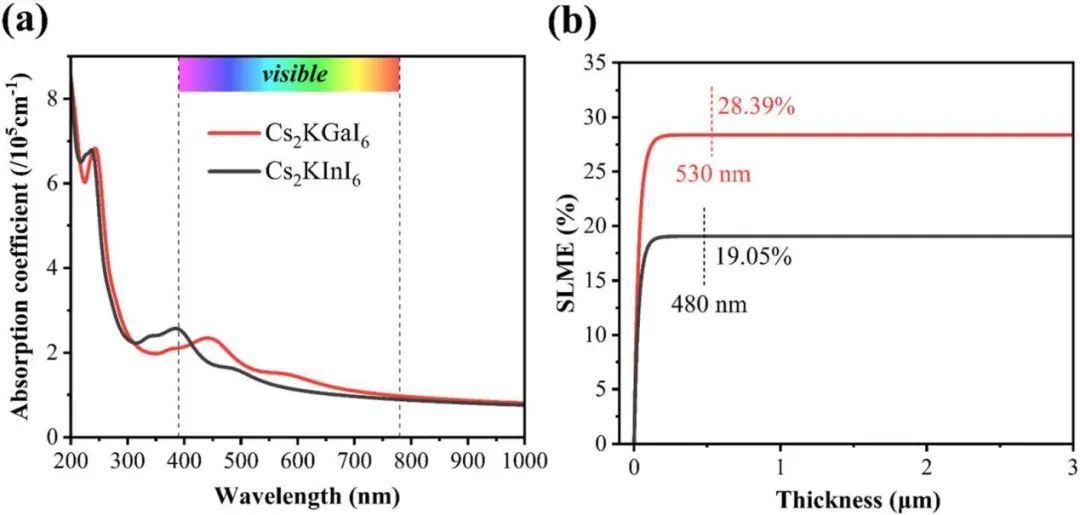
图5 光吸收谱及SLME图
结论与展望
从1026个化合物中,作者根据适当的结构因子、正分解能和合适的直接带隙等标准,精细地选择了钙钛矿Cs2KMI6(M = Ga,In)。还利用原始原理方法从理论上研究了这两种钙钛矿的热稳定性、光电性能和缺陷效应。根据结构因子、分解能和稳定化学势区域的计算,评估了这些化合物的结构稳定性。得出结论,K-I键具有共价性质,而M-卤素键和Cs-I键是离子键。此外,这些化合物的计算有效质量相对较小,从而有利于载流子的输运。这些基于第一性原理方法的光电特性可以集成到数值模型中,以预测未来的电流-电压曲线。此外,作者预测了不同类型的深层缺陷,这可能会导致陷阱和中心重组,从而导致器件性能下降。这些理论结果有助于研制和设计新型用于高效光电器件的无铅双钙钛矿,以及调整其光电性能的缺陷工程。
文献信息
Qi, F., Lv, X., Song, J., Fu, X., Meng, L., & Lu, C. Z. (2023). Optoelectronic performance of perovskites Cs2KMI6 (M= Ga, In) based on high-throughput screening and first-principles calculations. Physical Chemistry Chemical Physics.
htt-ps://doi.org/10.1039/D3CP00732D

















 1040
1040

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









