











研究背景
分子和原子物种在固态材料表面上的化学吸附是化学、物理学和材料科学中的一个中心概念。识别控制化学吸附强度的关键表面和吸附质性质的能力对于理解表面科学中的化学过程至关重要。在多相催化反应中,中间体与表面之间的键合强度提供了关于催化活性和选择性的决定性信息。尽管有几十年的研究,但仍然缺乏能够预测具有活性位点分辨率的复杂催化剂结构和组成的吸附能的可解释建模方法。
SLAC国家加速器实验室Frank Abild-Pedersen等人推导了一个完全基于物理参数的模型,准确地描述了O、N、Cu、Li(MAE为0.13 eV)的吸附能。变量很容易计算。基于有限的DFT训练数据,对特定吸附质和吸附部位的模型参数进行了优化。考虑了吸附诱导的吸附位点的变化,以及这些变化如何与位点周围化学环境的变化相互作用。这些变化明确地导致偏离典型的吸附能的线性行为与电子结构描述符。该模型是在小扰动的极限下推导出来的,因此,只有在考虑对给定位点基序的微小变化时才有效。通过对一个具有代表性的位置基序的适当校准,该方法可以完全推广到整个过渡金属系列和多组分系统。该方法为发现由复杂的多金属材料组成的改进催化剂提供了一条很有前途的途径。
计算方法
本文使用VASP软件采用PAW赝势、自旋极化方法进行,用于表示核心电子态,始终采用电子交换和相关能量的广义梯度近似(GGA)(RPBE)函数形式。表面计算的截断能值为600 eV。所有模型的几何优化采用的力收敛准则为0.01eV Å−1。模型结构由面心立方金属结构Au, Ag, Cu和Pt创建。在由6层面心立方金属原子构成的2×2(111)表面上研究近表面合金(NSAs)的模型表面。几何优化后,使用12×12×1和6×6×1K点网格进行单点计算。从这项研究中获得的所有结构和能量都上传到催化Hub90,并可以通过https://www.catalysis-hub.org/ publications/SainiElectronic2022直接访问。结果与讨论因为广泛和无结构分布的电子态的过渡金属和过渡金属合金,相互作用可以假定近似常数,因此,吸附表面之间的差异略有不同表面将完全由d状态之间的差异和重整化吸收状态。在下面,将分别使用ε+和ε−来定义成键态和反成键态(图1b,c)。
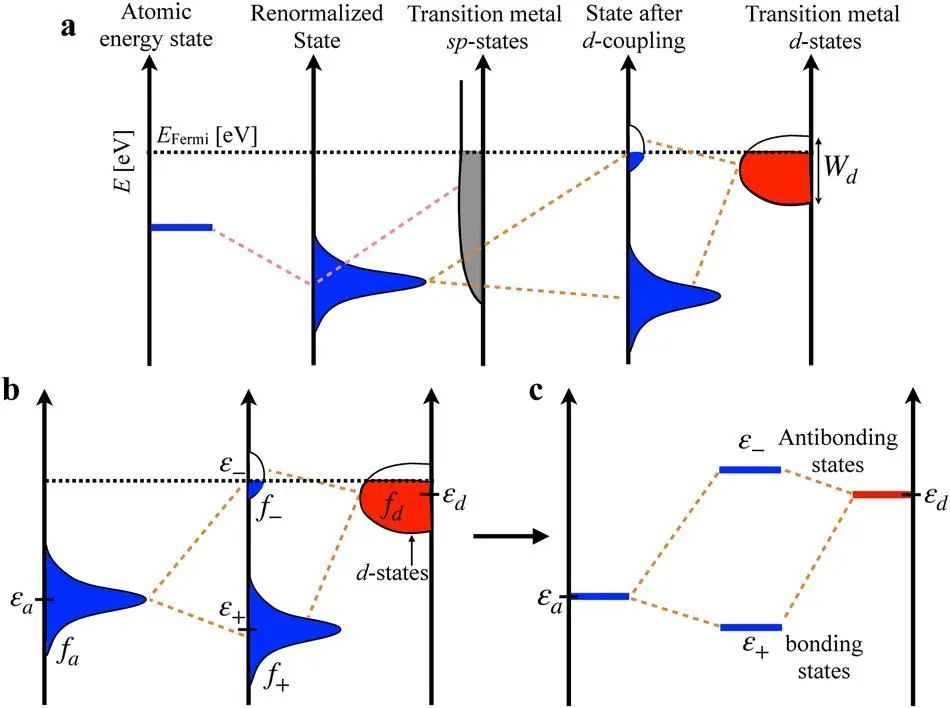
图1 吸附质与过渡金属表面态之间的相互作用
在图2a中,只有O的结合与V2有明显的相关性,未能描述N和CH的结合,则f-==f的结果部分成立。则探究为何在这两种情况下不成立由图2b、c可知是由于涉及金属d态和吸附剂上重整态之间的杂化。从以上可以看出,在确定化学吸附趋势时,吸附质的影响不容忽视。因此,作者更普遍地探索了在何种情况下,吸附物诱导的吸附位点及其周围环境的扰动会影响表面的相互作用。
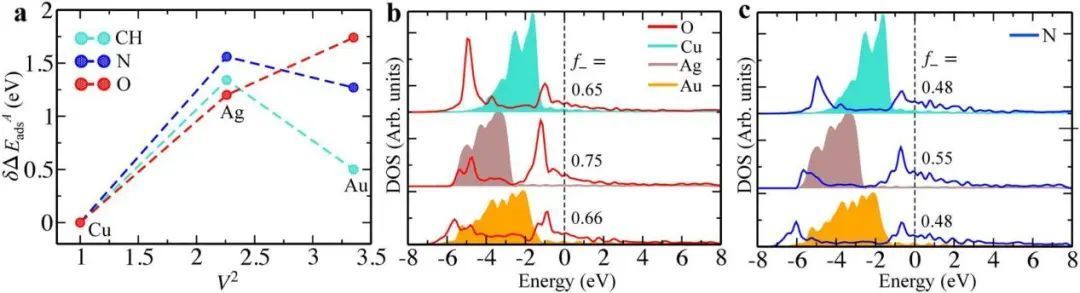
图2 在纯过渡金属表面上的化学吸附 a图 DFT计算了O、N和CH对Cu(111)、Ag(111)和Au(111)的吸附能作为耦合矩阵元素(V2)的函数。b图、c图分别为O(红线)和N(蓝线)的p投影态密度
假设吸附质对金属位点的影响在化学吸附过程中起着不可忽视的作用。为了验证,进行电子结构分析。表明当电子结构参数εd和Wd(d带宽度)发生变化时,吸附位点存在大量吸附效应。在图3a中,绘制了位点稳定性,表明,它与εd的变化以及CN的变化相关,即δεd∝δCN∝δBEM。CN最高的位点的εd最低,导致位点稳定性增加,从而与吸附质的相互作用减弱。在图3b中,金属吸附质配合物的吸附能与位置稳定性之间的相关性与CN变化时的线性相关性有所偏离。这种差异只能是吸附质影响吸附部位的局部电子结构的结果。
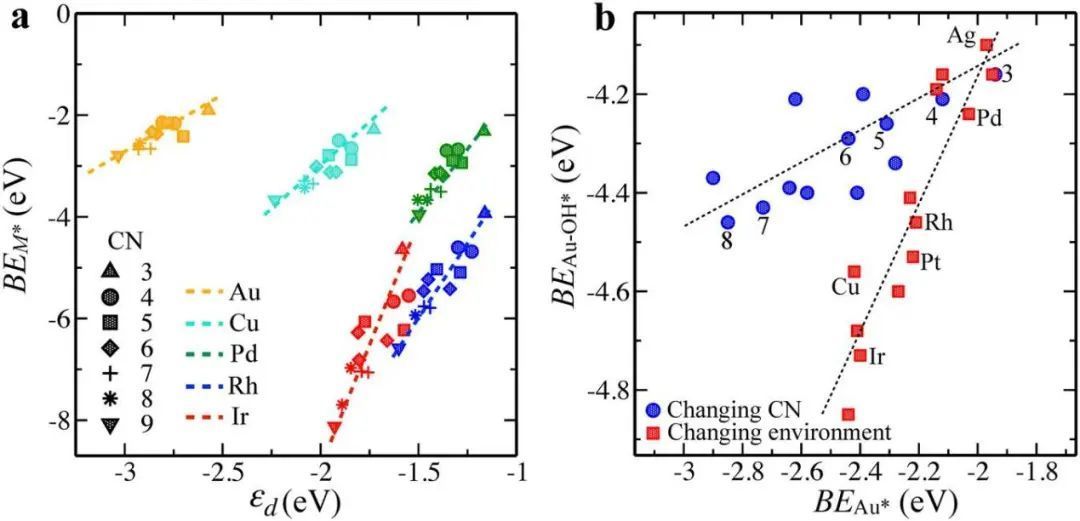
图3 位点稳定性随d带中心、配位数和站点环境变化的变化。a图 金属原子稳定性作为面心立方金属结构(100)、(111)和(211)表面原子数变化的表面上d带中心(εd)和配位数(CN)变化的函数。b图 纯金和金表面合金的金属吸附配合物的结合能作为金原子结合能的函数。
在图4a中,只考虑了衬底中的电子态如何影响吸附质上的电子能级,这种理解表面相互作用的最低阶近似已经应用于大多数化学吸附模型。在所有情况下,这种近似导致的模型只依赖于与金属表面严格相关的参数,即状态的d带密度、d带中心、表面加权配位数和位置稳定性,并且都与等式(1)相关。图4b中引入的二阶项由吸附质扰动的表面与原始金属表面的差给出。已知,内聚能差可以用电子性质来表示。如果假设d带有一个简单的矩形形状,宽度为Wd,其中Wd与耦合矩阵元素V成正比,内聚能可以写为:
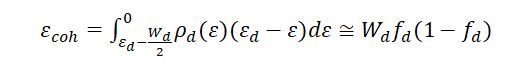
为最接近吸附位点的金属原子的d态的填充,ρd(ε) 为d态的能量密度函数(态密度,DOS),因此,对于金属体系,结合能(εcoh-)取决于ρd(ε)分布中的电子数及其随能量(ε)的变化。特定的吸附效应取决于的性质。因此,内聚能的变化与吸附位点的) d带结构的变化成正比,也取决于相邻原子的) d态的填充程度。
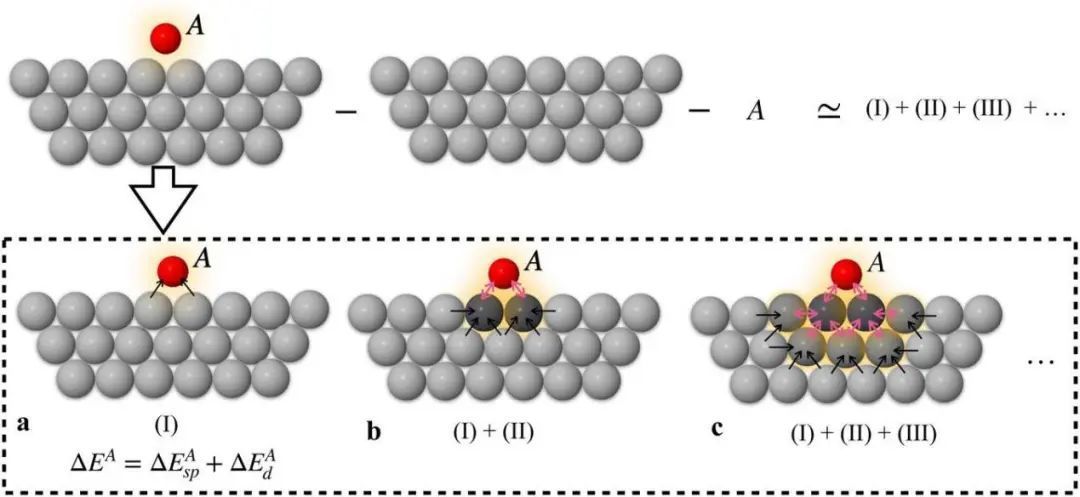
图4 吸附质-表面相互作用示意图。a图 显示零阶近似键。b图、c图分别引入一阶效应和二阶效应
NSAs引入了发生在材料亚表层的成分变化。因此,吸附位点是固定的,吸附相对于参考状态的所有变化都可以完全归因于围绕物相对于参考状态引起的电子结构的微小扰动。考虑了NSAs,其中第二层原子跨越了3d、4d和5d过渡金属系列的全部元素范围,如图5a所示。在图5b中,大多数情况下,d带中心相对于费米能级向下位移,因此f=1保持不变,根据d带模型,通过等式(10)的第一项没有额外的键合为预期值。以及随着d带中心的下降,氧的结合能变得更强,证明了模型推导的正确性,明确考虑了反键态的填充。
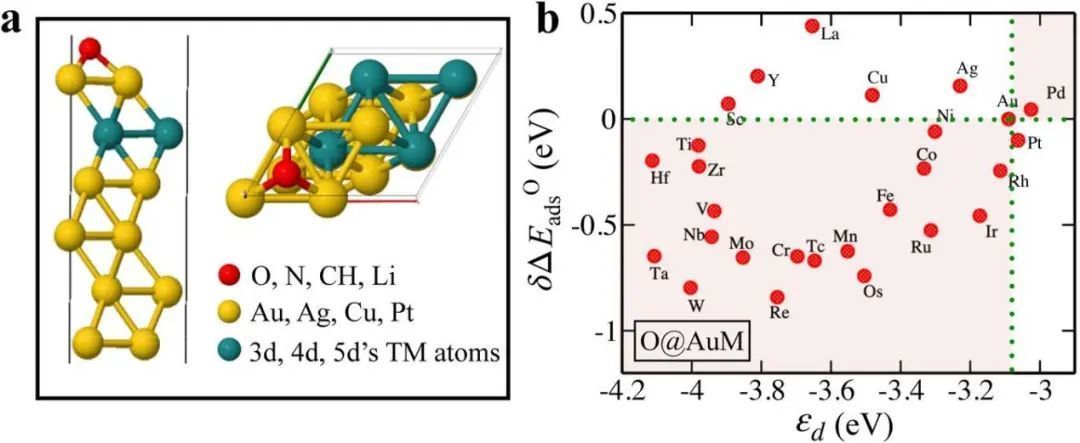
图5 NSAs上的化学吸附作为d带中心的作用;a图在nsa上吸附的顶部和侧视图。b图O在Au NSAs上的吸附能随d带中心(εd)的变化。
在图6a中, fd中所有吸附物的二阶行为与等式(18)中引入的第二项一致。根据吸附质的不同在元素周期表中向左移动时,重整化的能级,εa被推向费米能级,也就是说,当清空一个原子轨道时,f−从O到Li逐渐减少,如图6b所示,以及如图6c从O移动到Li时,排斥力线性增加或减少。而且随着O、N、CH和Li的变化,斥力程度会增加。这种效应在图6a中很明显。
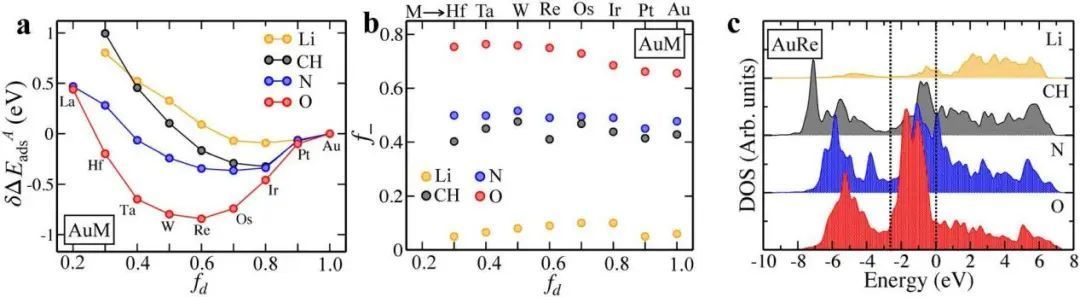
图6 化学吸附作为fd的函数。
a图DFT计算了O、N、CH和Li在第二层含有5d金属的金NSAs上的吸附能作为fd的函数。(所有的能量都是相对于纯金的)。b图 吸附物的反键态的填充因子(f−)作为fd的函数。c图 吸附在AuRe NSA上的O、N、CH和Li的DOS。如图7所示,对于由于吸附导致的自旋和自旋跃迁起作用的情况,对稀土金属NSAs的影响要大得多。这些结果表明,在非铸造体系上,自旋猝灭被d态屏蔽。当考虑NSAs时,主机是具有非填充d态的金属,如Pt,它引入了额外的复杂性。图7d-f中Pt NSAs的结果与Au NSAs的结果有很大差异。可以观察到,当在不同的d系列中嵌入过渡金属时,Pt NSAs上的相对结合能也表现出抛物线行为,而抛物线行为对O最强,当观察N和最后观察CH时减小。对于Au和剩余的稀土金属NSAs,线性项在N和CH情况下变得更占优势,对于Pt也是如此(图7e,f)。此外,由于f位点d =1,对于不同的吸附物,f−要小得多,这表明计算模型可以参数化,仅从裸露NSAs的电子结构信息来预测化学吸附能。

图7 对Au和Pt基NSAs的化学吸附变化。a-c图 DFT计算了O、N和CH(ΔEA ads)在Au NSAs上的吸附能的变化。d-f图 分别表示O、N、CH对Pt NSAs的吸附能相对于纯Pt的变化
对于给定的位置和吸附剂,系数是常数,相对于参考系统的变量是独立于吸附剂的。因此,一旦系数被拟合,可以很容易地获得训练集之外的系统的结合强度。通过图8a,很明显,与其他NSA相比,Pt数据点在奇偶性线上明显更分散,特别是涉及早期过渡金属的合金。对于在模型表达式中使用εd作为自变量相关的偏差,有一个潜在的物理解释。然而,当接近早期的过渡金属时,它开始弯曲。因此,吸附能不能与εd保持线性相关关系,如图8c所示。这种效应可以通过引入一个包含d带结构信息的参数来弥补,因此与费米能级的联系更强。由εd+Wd/2近似给出的上d带边是一个更好的描述符,因为它能有效地识别出反键态的位置(图8d)。

图8 利用该模型预测吸附能 a图,b图利用方程式中的模型预测了O、N、CH在Au、Ag、Cu和Pt NSAs上的吸附能。c图,d图表示Pt NSAs上的O吸附能分别随εd和εd+Wd/2的变化。混合表面和合金内部的可转移性
该模型要作为催化材料设计的指导,它需要能够处理不同结构和组成的通用多金属体系。下面将讨论扩展模型所需的修改。图9a所示的趋势在fd中明显为二阶,但与NSAs相反,表面合金的吸附位点稳定,说明表层内的相互作用与与下层的相互作用有显著差异。这些原子将与周围的表面和内部原子相互作用,以降低整体表面能,如图9b和d所示。如果假设(111)表面上一个原子上的剩余电子态被推到相邻的表面层原子上,那么就会在一个完全对称的表面上引入原子间电荷密度的差异。因此,密度较高的原子必须移动以补偿额外的电荷,从而导致表面弯曲,如图9c(i)所示。另一方面,如果额外的电荷被推向地下的原子,其结果将是表层原子的收缩/膨胀,如图9c(ii)所示。证明了表面和客体原子与吸附位点的相互作用会不同。

图9表面合金上的化学吸附图。a图显示了金表面合金(SAs)上的吸附能随fd的变化。b-d图说明了表面和地下层原子内电荷转移的影响。
为了测试模型表达式准确性,得出图10b所有计算和建模的数据点都遵循奇偶性线。因此,这证实了本文推导出的概念框架,因此可以用于预测和解释在广泛的合金表面上的化学吸附。模型仅基于四个参数(βd,βf d;1,βf d;2,γ)与单纯基于数据驱动方法(MAE:0.10-0.25eV)的模型具有相似或更好的性能。D带中心与带宽的有效描述有望应用于催化剂的设计,具有更好的物理洞察力和可解释性。
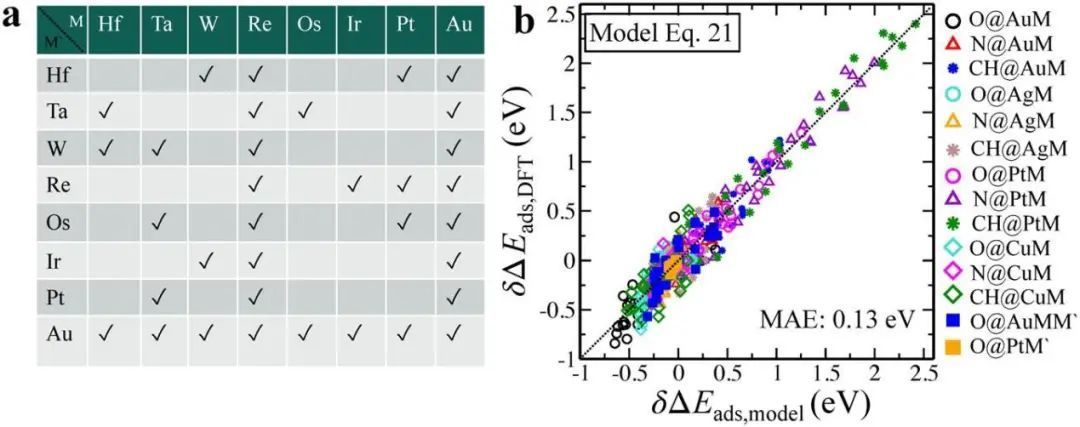
图10 奇偶性图显示了我们的模型在混合表面合金上的可转移性。a图描述了随机选择的Au基混合表面和合金内部,b图由等式中的模型预测的化学吸附能
结论与展望
在这项工作中,作者提出一个基于物理学的化学吸附模型,它包含了吸附剂引起的表面-位点的扰动。通过基态电子因素和吸附物的特性来解释这一点,即d-填充分数(fd)、d-带中心、dstates的宽度、反键合状态的填充以及重新规范化的吸附物状态的位置。该方法已被推广到整个系列的过渡金属和多组分系统,可以用来推动该领域的发展,以指导具有理想催化性能的复杂合金的工程。
文献信息
Saini, S., Halldin Stenlid, J., & Abild-Pedersen, F. (2022). Electronic structure factors and the importance of adsorbate effects in chemisorption on surface alloys. npj Computational Materials, 8(1), 163.
ht-tps://www.nature.com/articles/s41524-022-00846-z















 2762
2762











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









