










成果简介
合成气选择性转化为高附加值醇(含两个或两个以上碳原子),特别是转化为一种特定的醇,是一个非常有趣的问题,但仍然具有挑战性。
中国科学技术大学路军岭教授、刘进勋教授、王恒伟副研究员等人发现,通过选择性地将高度分散的FeOx构建在四方氧化锆负载的超细筏状Rh团簇上,可以实现FeOx-Rh-ZrO2双界面的原子紧密组装,从而实现合成气到乙醇的高效串联转化。在CO转化率高达51%的情况下,乙醇在氧合物中的选择性达到90%,乙醇的时空产率高达668.2 mg gcat-1 h-1。原位光谱表征和理论计算表明,Rh-ZrO2界面通过甲酸途径促进解离CO活化成CHx,而邻近的Rh-FeOx界面通过非解离CO插入、加速随后的C-C耦合。因此,这些具有互补功能的原子尺度接近的双界面协同促进乙醇的特异性形成,并以串联方式显示出卓越的生产效率。
中科大博士毕业生李尚和博士研究生冯丽为该论文共同第一作者。
相关工作以《Atomically intimate assembly of dual metal–oxide interfaces for tandem conversion of syngas to ethanol》为题在《Nature Nanotechnology》上发表论文。
图文介绍

图1 选择性ALD合成FeOx-Rh-ZrO2双界面催化剂示意图
本文首先使用溶胶-凝胶法合成了t-ZrO2载体,然后使用初湿浸渍(IWI)法制备了Rh/ZrO2催化剂,Rh含量为3.3 wt%。HAADF-STEM和EDS图谱显示,Rh在t-ZrO2上高度分散,团簇大小为1.1±0.3 nm,并伴有少量孤立的Rh原子。接下来,在150℃下,采用原子层沉积法(ALD)将FeOx选择性沉积在Rh团簇上,制备FeOx-Rh-ZrO2双界面催化剂。
为了最大限度地减少FeOx ALD对Rh-ZrO2界面的堵塞,首先将Rh/ZrO2催化剂暴露于乙二醇(EG)中,通过形成稳定的烷氧化物来钝化ZrO2上潜在的ALD成核位点,即羟基,同时保持Rh位点完整。然后,FeOx的ALD在EG预处理的Rh/ZrO2上进行不同次数的循环,以调整FeOx在Rh团簇上的覆盖范围(xFe-Rh/ZrO2,其中x表示ALD循环次数);最后,完全去除EG,重新暴露Rh-ZrO2界面。

图2 催化合成气制乙醇的性能
在相同条件下(260℃,2.5 MPa,H2:CO=3:1),对这些Rh催化剂的催化性能进行了评价。如图2a所示,Rh/SiO2的CO转化率仅为2.1%,乙醇的STY为3.8 mg gcat-1 h-1。相比之下,Rh/ZrO2的CO转化率为14.6%,乙醇的STY为46.1 mg gcat-1 h-1,而乙醇选择性仍然限制在54.6%左右。相比之下,Rh/Fe2O3在氧合物中表现出更高的STY(112.6 mg gcat-1 h-1),CO转化率为16.3%,乙醇选择性为66.0%,这表明Rh-FeOx界面是有活性的,更有选择性的C-C耦合形成EtOH。然而,这三种催化剂都不可避免地产生了相当数量的甲醇(图2a)。
具有FeOx-Rh-ZrO2双界面的xFe-Rh/ZrO2催化剂的催化性能得到显著提高。动力学测量进一步表明,6Fe-Rh/ZrO2的表观活化能为~87.2 kJ mol-1,明显低于Rh/ZrO2 、Rh/Fe2O3和Rh/SiO2(图2b),验证了更高的本征活性。同时,6Fe-Rh/ZrO2在氧合物中的甲醇选择性仅为7.8%。将WHSV从60000 ml gcat-1 h-1降低到9000 ml gcat-1 h-1,即使在CO转化率为50.9%的情况下,乙醇在含氧化合物中的选择性也保持在88%以上(图2c)。值得注意的是,在温和的反应条件下,6Fe-Rh/ZrO2催化剂在CO转化率为31.5%的情况下实现了668.2 mg gcat-1 h-1的乙醇生成率,在含氧化合物中选择性为~90%,远远优于目前报道的最先进的催化剂(图2d)。
更重要的是,6Fe-Rh/ZrO2还表现出至少200小时的优异稳定性,没有发生任何Rh聚集,活性或选择性也没有明显下降(图2e),这表明它具有巨大的实际应用潜力。
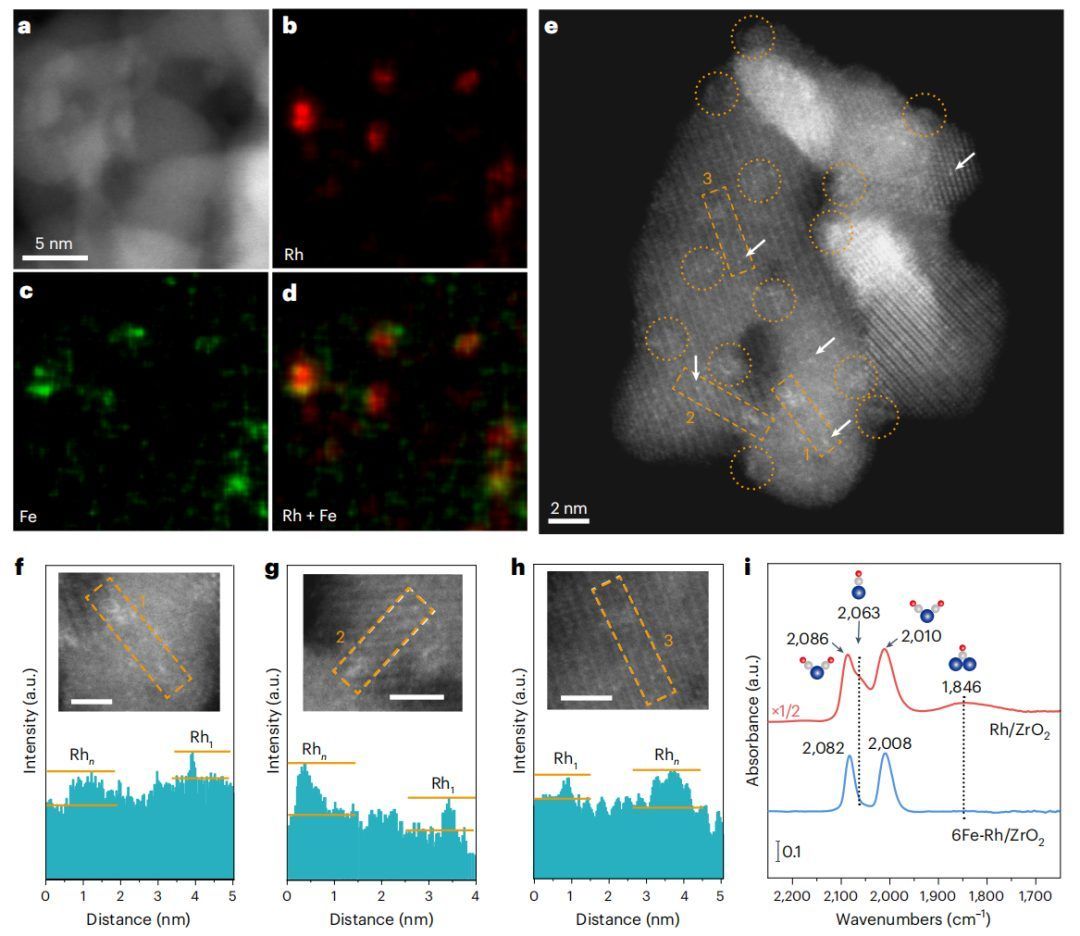
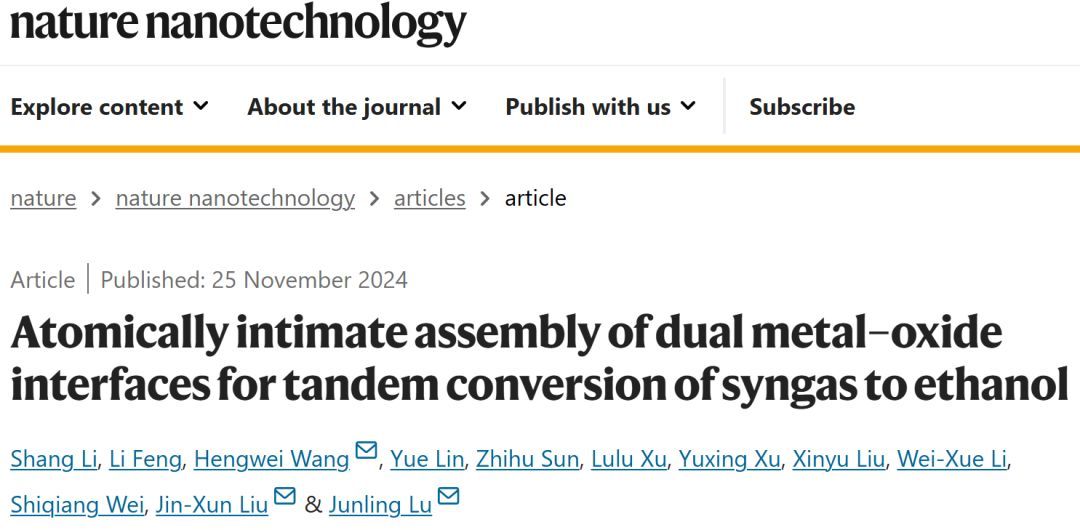
图3 6Fe-Rh/ZrO2的形貌
STEM-EDS元素图谱显示,6Fe-Rh/ZrO2中的Fe和Rh信号相互强烈重叠,证实了FeOx在Rh团簇上的选择性沉积(图3a-d)。FeOx包覆的Rh团簇平均直径为1.2±0.2 nm(图3e),其强度也仅略高于孤立的Rh原子(图3f-h),表明FeOx修饰后保留了筏状结构。在ZrO2上的FeOx-Rh具有高度分散和原子厚度的特点,不仅可以最大限度地利用Rh原子,而且可以使Rh-ZrO2和Rh-FeOx界面在原子尺度上接近。
对于FeOx包覆的Rh/ZrO2催化剂,随着FeOx的ALD循环的增加,6Fe-Rh/ZrO2上线性和桥键CO峰的强度比双羰基CO峰的强度衰减得更快(图3i),表明了Rh-FeOx界面发生调整。
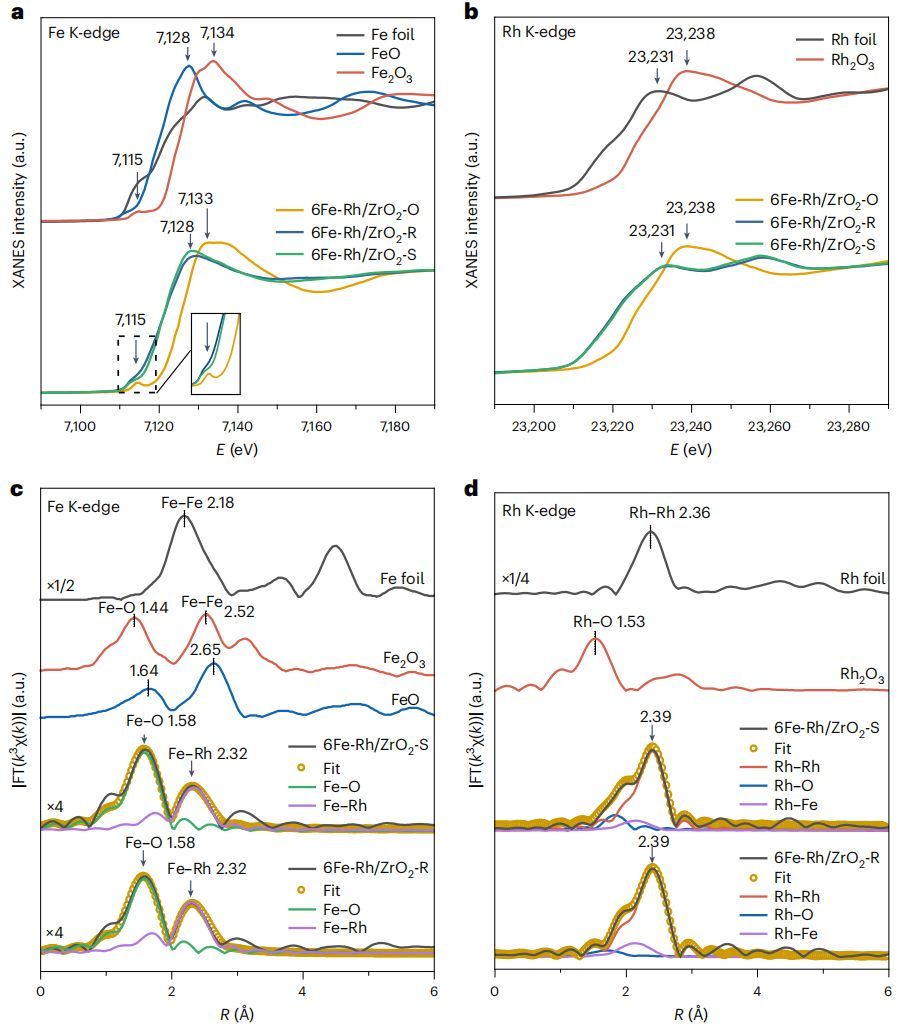
图4 原位XAFS测试
为了确定实际反应条件下Rh-FeOx界面的结构,进一步进行了原位XAS测量。在Fe的K边,原始样品(6Fe-Rh/ZrO2-O)在400℃、H2还原后(6Fe-Rh/ZrO2-R),7115 eV处对应Fe3+氧化物1s-3d跃迁的前边峰随着吸收边的下移而消失,甚至略低于FeO参考样品(图4a)。切换到260℃、合成气氛围(6Fe-Rh/ZrO2-S),前边峰重新出现,吸收边与FeO相似,表明Feδ+(0<δ<2)轻微再氧化为Fe2+。在Rh的K边,6Fe-Rh/ZrO2-R和6Fe-Rh/ZrO2-S的吸收边与Rh箔相似,说明在反应条件下,大部分Rh处于Rh0金属态(图4b)。
EXAFS光谱显示,与6Fe-Rh/ZrO2-O相比,6Fe-Rh/ZrO2-R和6Fe-Rh/ZrO2-S的Fe-O峰均明显减弱(图4c),配位数(CN)为1.7-2.3。同时,Fe-Fe配位峰(>2.5 Å)的缺失说明在反应条件下,大部分Fe呈原子分散。在Rh的K边,6Fe-Rh/ZrO2-R和6Fe-Rh/ZrO2-S都显示出一个宽的Rh-Rh配位峰,位于2.39 Å, CN为~6.5(图4d),与裸Rh/ZrO2相似,这与Rh团簇的“筏状”结构非常吻合。

图5 CO加氢的原位DRIFTS研究
作者进行了DRIFTS测量,以深入了解双界面协同作用。在260℃引入CO后,在Rh/SiO2上观察到两个CO峰在2055 cm-1和1935 cm-1处(图5a)。相比之下,在t-ZrO2上观察到强甲酸峰以及宽的羟基峰(以3723 cm-1为中心)。有趣的是,这些甲酸盐峰在Rh/ZrO2和6Fe-Rh/ZrO2上变得更强,CO峰在2044 cm-1和1830 cm-1处也变得更强,这表明甲酸盐更有利于在Rh-ZrO2界面形成。
将CO转换为H2,Rh/SiO2上的CO峰下降缓慢。对于Rh/ZrO2,CO峰在H2中迅速下降,而HCOO*、CH3O*(2925 cm-1)和气态CH4(3015 cm-1)的峰都在早期略有上升,然后随着时间的推移逐渐下降(图5b、d)。显然,CO在Rh-SiO2界面或Rh金属表面上的加氢都很困难,但很容易通过甲酸途径在Rh-ZrO2界面上发生。
在6Fe-Rh/ZrO2中,将CO转换为H2可以更快地降低HCOO*(图5c、d)。与此同时,CH3O*在初期没有任何积累,而是瞬间减少,与Rh/ZrO2形成鲜明对比(图5d)。考虑到HCOO*在Rh-ZrO2界面上加氢生成CH3O*或CHx*的竞争,这种抑制CH3O*的形成意味着反应路径向HCOO*→CHx*转变。这种转变可能是由于在邻近的Rh-Fe1O2界面上,通过C-C与吸附的CO*耦合形成EtOH加速了CHx*的消耗,并且6Fe-Rh/ZrO2上CO*的消耗比Rh/ZrO2上更快。
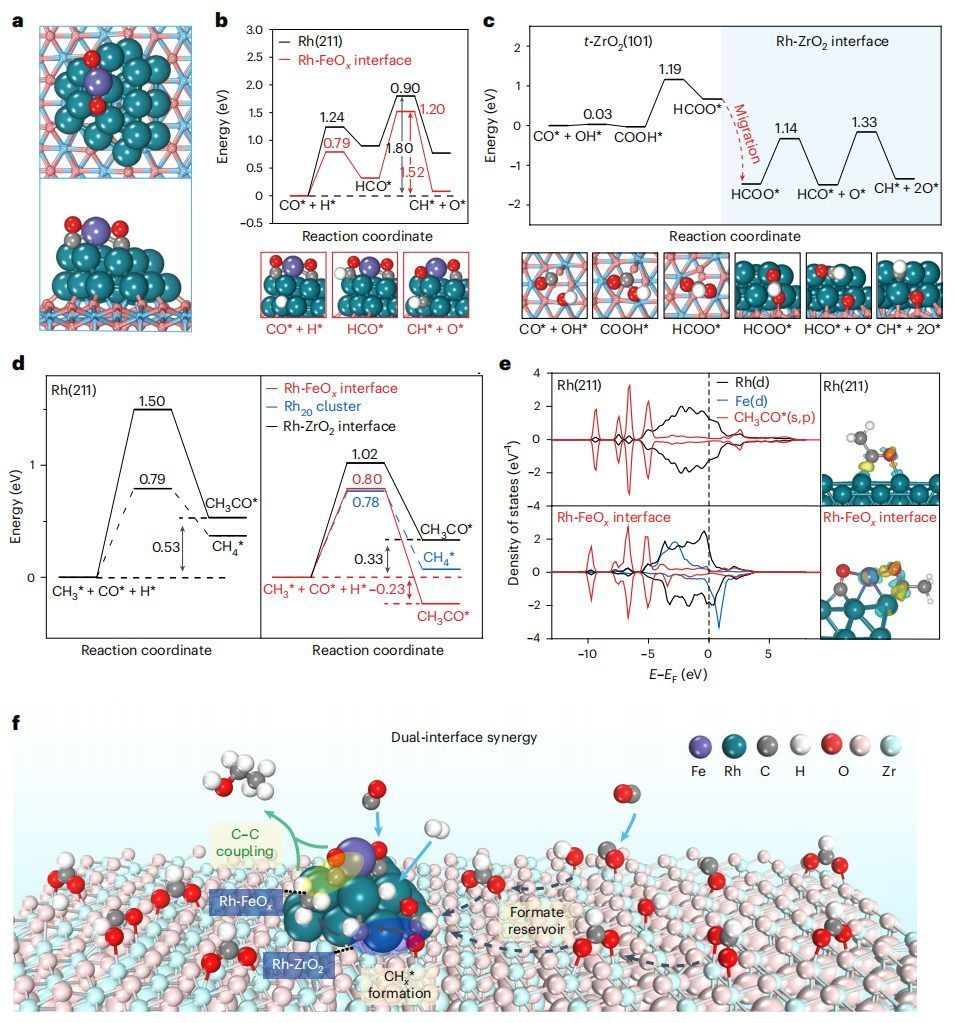
图6 DFT计算
本研究进一步进行DFT计算,以更深入地了解双界面协同作用。如图6a所示,采用含有羟基和VO的t-ZrO2最稳定(101)面作为载体,构建双界面模型。在反应条件下,对单个Fe原子在Rh20基团上与多种可能的O、OH和CO配体进行测试后,发现分离的Fe1Ox优先结合在Rh20基团的Rh桥位上,Fe原子配位到两个Rh原子和两个倾斜的CO分子(表示为Fe1(CO)2/Rh20/t-ZrO2(101))。
CO活化通常被认为是该反应在Rh催化剂上的速率决定步骤。在Rh(211)上,发现通过HCO*中间体的氢辅助CO活化比直接解离更有利,但仍然明显吸热0.77 eV,具有1.80 eV的高势垒(图6b)。在Rh-FeOx界面上,氢辅助CO活化非常容易,反应能降低为0.08 eV,势垒为1.52 eV。这是由于HCO*和O*中间体的吸附增强。在t-ZrO2(101)上,直接解离和氢辅助解离在热力学上都是不利的。
相比之下,发现强吸附的CO*(ECO=-1.92 eV)很容易与OH*反应生成COOH*,然后以1.19 eV的中等势垒转化为HCOO*(图6c)。接下来,HCOO*放热迁移到Rh-ZrO2界面,因为吸附能从-1.91 eV增加到-3.47 eV,这与DRIFTS观察到的Rh/ZrO2和6Fe-Rh/ZrO2上的HCOO*强度比裸t-ZrO2强得多一致。然后,HCOO*在Rh-ZrO2界面上依次解离成HCO*和CH*,势垒最大为1.33 eV。因此,与Rh金属和Rh-FeOx界面相比,Rh-ZrO2界面对CO转化为CHx*更有利,其中羟基化的t-ZrO2载体作为HCOO*储层促进反应。
对于C-C耦合,CO*插入CH3*是Rh(211)、Rh-FeOx和Rh-ZrO2界面最有利的路径。尽管如此,在Rh(211)上,CO*插入到CH3*中形成CH3CO*(EtOH的关键中间体)远不如CH3*直接加氢生成甲烷(图6d),这使得在Rh/SiO2上观察到的甲烷是Rh金属表面上的优先产物。然而,在Rh-FeOx界面上,由于轨道杂化增强和Fe向CH3CO*的大量电荷转移,CH3CO*在Rh(211)上的吸附要比CH3CO*强得多(图6e)。因此,在Rh-FeOx界面上,CO插入CH3*形成CH3CO*变成了-0.23 eV的放热反应,同时具有0.80 eV的低势垒,这在能量上比CH3*的加氢反应更有利。
为了在合成气转化过程中获得高的EtOH产率,确保两个关键的基本反应步骤的平衡至关重要:CO活化到CHx*和随后通过CO*插入到CHx*中的C-C偶联,这两个步骤都是高效的。计算结果表明,双界面协同作用实现了串联反应过程,即Rh-ZrO2界面可以有效活化CO*通过甲酸路径形成CHx*,ZrO2载体作为甲酸储层;然后,形成的CHx*很容易与另一个CO*分子耦合,在邻近的Rh-FeOx界面上形成CH3CO*中间体,从而协同促进特异性EtOH的形成(图6f)。
文献信息
Atomically intimate assembly of dual metal–oxide interfaces for tandem conversion of syngas to ethanol,Nature Nanotechnology,2024.

















 1291
1291

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









